|
||||
|
|
||||
|
||||
|
|
||||
|
Lipoproteínas y riesgo cardiovascular Dres.
Raúl Gamboa (*) EL TRANSPORTE DE LOS LIPIDOS Y EL RIESGO CARDIOVASCULAR Transporte de Lípidos Exógenos (dietario):
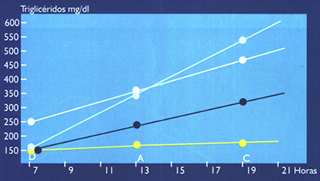
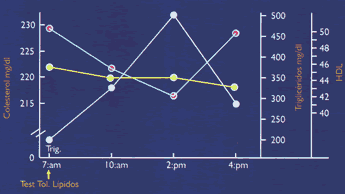
Cuando se ingieren alimentos conteniendo grasas (triglicéridos) y colesterol, estos lípidos son absorbidos dentro de células del intestino delgado como ácidos grasos y colesterol. Dentro de las células mucosas se produce la re-esterificación de los ácidos grasos a triglicéridos, y del colesterol a ésteres de colesterol. De esta forma estos lípidos hidrofóbicos se incorporan al núcleo del naciente quilomicrón, iniciándose así la vía del transporte exógeno del colesterol hacia el hígado. El quilomicrón viaja inicialmente hacia el hígado con el 90% de su peso constituido por triglicéridos. En el camino es hidrolizado por la lipasa lipoproteíca (LPL), desembarcando sus triglicéridos, y convirtiéndose en una lipoproteína más pequeña, el remanente del quilomicrón, relativamente rico en ésteres de colesterol, y que será captado por los receptores hepáticosE2(1-4), (ver Figura 1). Relación entre Transporte Exógeno y el Riesgo Cardiovascular: Si el transporte y el metabolismo de las lipoproteínas derivadas del intestino es normal, los triglicéridos de los quilomicrones serán entregados a los adipocitos y células musculares como ácidos grasos, y el colesterol será entregado al hígado. Sin embargo, si el transporte y el metabolismo son anormales, los quilomicrones pueden desempeñar un importante rol en el proceso ateroesclerótico. Diferentes investigaciones revelan que los remanentes del quilomicrón son altamente aterogénicos(5), y que en presencia de disfunción endotelial, es decir en presencia de factores de riesgo, son rápidamente captados por las células endoteliales y macrófagos, dando origen a las células espumosas, la más temprana lesión de la placa ateroesclerótica. Si los niveles plasmáticos post-prandiales de quilomicrones o sus remanentes son muy elevados (lipemia post-prandial), las probabilidades de entregar colesterol a la pared arterial se incrementan notablemente(5), (ver figura 2 y figura 3) (6). Por supuesto que la ingestión prolongada y abundante de alimentos, con alto contenido de grasas saturadas y colesterol, elevará agudamente los niveles de remanentes. Otras situaciones también pueden prolongar el tiempo de residencia plasmática de los quilomicrones y sus remanentes:
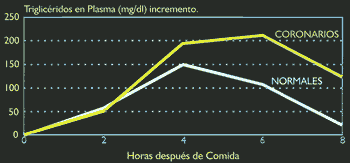
a) reducción en la actividad de la LPL, tal como ocurre en la diabetes mellitus, o cuando hay un desbalance entre sus proteínas constitutivas, apoC11 (activadora de LPL) y apoC-III (inhibidora de la LPL); b) el polimorfismo de su apoproteína E2, es causa de la disbetaliproteinemia, alteración genética altamente aterogénica en la cual la apoE mutante del remanente no es reconocida por el receptor hepático, constituyéndose en el paradigma del metabolismo defectuoso de los remanentes del quilomicrón. Recientes estudios confirman que los pacientes coronarios presentan lipemias postprandiales mayores y más tardías que los individuos sanos `, (ver figura 4). Transporte de Lípidos EndógenosEl sistema de transporte endógeno conduce lípidos desde el hígado hacia los tejidos periféricos, a través de las lipoproteínas VLDL, IDL, y LDL, y desde los tejidos periféricos de retorno hacia el hígado, a través de las HDL (9) (ver Figura 1).
El tamaño de la VLDL depende principalmente de la cantidad de triglicérido disponible. Así, grandes VLDL ricas en triglicéridos se presentan en obesidad, diabetes mellitus, y consumo de alcohol. Una vez en el plasma, las VLDI- son hidrolizadas por la LPL en la misma manera que los quilomicrones, transformándose en pequeños y densos remanentes capaces de ser captados por receptores hepáticos, o reducirse aún más convirtiéndose en las IDL que darán origen a las LDL. Una vez formada la LDL, su biodisponibilidad plasmática depende esencialmente de la capacidad de los receptores presentes en los hepatocitos (10). Se considera que el 75% del catabolismo de las LDL es efectuado vía receptores hepáticos, el resto es realizado a través de mecanismos no específicos(10).
Las HDL son sintetizadas y secretadas por el hígado e intestino, así como del resultado del catabolismo de quilomicrones y VLDL. Las HDL son las lipoproteínas encargadas del transporte reverso, mecanismo por el cual el exceso de colesterol tisular es retornado hacia el hígado (Figura 5)(9). Relación entre el Transporte Endógeno y el Riesgo Cardiovascular:
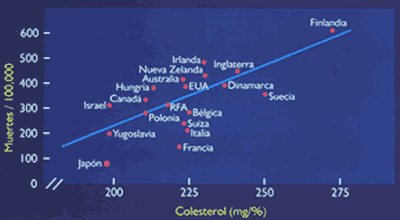
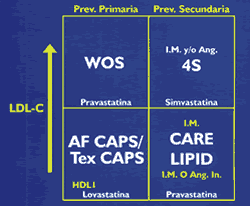
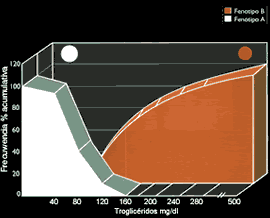
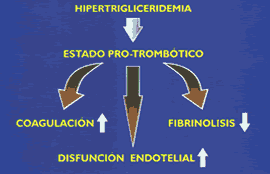
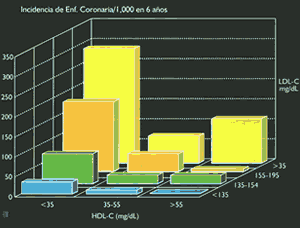
Los incrementos plasmáticos del colesterol, y su principal transportador, las LDL, han demostrado una clara relación con el riesgo de desarrollar enfermedad coronaría (10) (Figura 6) y (Tabla 1). De otro lado, mega-estudios de prevención primaria y secundaria, utilizando estatinas, drogas inhibidoras de la síntesis del colesterol hepático, han demostrado significativas reducciones en la morbi-mortalidad coronaría, (Figura 7 y Figura 8). Sin embargo, es importante recordar que cuando se incuban LDL normales con macrófagos o con células musculares lisas, no es posible lograr la formación de células espumosas, el heraldo del proceso ateroesclerótico. Es evidente que se requieren alteraciones cualitativas en las LDL circulantes para que adquieran poder los aterogénico. Su modificacion por oxidación, glicosilación (en diabetes), agregación, atrapamiento por proteoglicanos, o incorporación en complejos inmunes, las convierten en la mayor causa de injuria al endotelio y a las células musculares lisas (18). Las secuelas biológicas de la oxidación de las LDL se muestran en la Tabla 2. Las modificaciones oxidativas no son exclusivas de las LDL, las mismas alteraciones pueden observarse en los remanentes de quilomicrón y VLDL, en las IDL, y Lp(a), razones por las que estas lipoproteínas también constituyen importantes factores de riesgo aterogénico (19). Otro aspecto importante de las LDL es la evidencia de la existencia de 2 sub- de esta lipoproteína: las del Tipo A, grandes y ricas en ésteres de colesterol, y las del Tipo B, pequeñas y enriquecidas en triglicéridos, y paradójicamente pobres en colesterol(20,21). Estas pequeñas LDL son altamente aterogénicas y están estrechamente relacionadas con el metabolismo de los triglicéridos (22)(ver Figura 9). Las hipertrigliceridemias de origen exógeno u endógeno, aunque no directamente relacionadas con la formación de la lesión ateroesclerótica, son contribuyentes de riesgo al causar importantes reducciones en el tamaño y densidad de las LDL y HDL (ver Figura 3 y figura 9), ambas alteraciones reconocidas como importantes factores de riesgo, Además, evidencias epidemiológicas, clínicas, y experimentales, sugieren que las hipertrigliceridemias constituyen un estado protrombótico por su acción pro-coagulante, antifibrinolítica, e injuriante endotelial (23)(Figura 10).
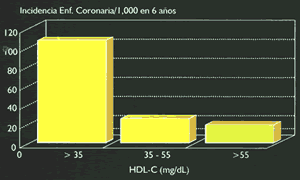
En contraste con el posible efecto aterogénico de las lipoproteínas participantes en el transporte exógeno y endógeno del colesterol hacia los tejidos, las concentraciones de HDL se relacionan inversamente con la incidencia de enfermedad coronaría (Fig.11), e influencian notablemente el efecto aterogénico del colesterol total y de las LDL (24) (Figura 12). Mas allá de su participación en el transporte reverso del colesterol, las HDL inhiben la migración monocítica y en consecuencia la formación del anión superóxido en los macrófagos, estimulan la formación de prostaciclinas induciendo por esta vía una mayor producción de óxido nítrico e inhibiendo la oxidación de las LDL (25). Se debe tener en cuenta que las concentraciones plasmáticas de HDL varían recíprocamente con la concentración de quilomicrones y VLDL, y directamente con la (6,7) actividad de la LPL.
En conclusión, los pacientes con enfermedad ateroesclerótica pueden presentar una de las siguientes anormalidades lipídicas: 1) lipemia post-prandial
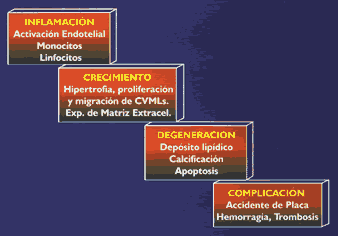
anormal, Como consecuencia de las anormalidades lipídicas antes mencionadas, hoy no se discute que la hipercolesterolemia es causa de enfermedad coronaria, y que la disminución del colesterol plasmático produce reducción en morbi-mortalidad coronaría. Sin embargo, se debe tener presente que la relación costo/efectividad de su tratamiento depende del riesgo absoluto, es decir del producto de: edad, sexo, enfermedad cardiovascular, y otros factores de riesgo. No obstante que las dislipidemias constituyen un indudable marcador de riesgo para enfermedad cardiovascular, es importante tener presente que sólo el 50 por ciento de los enfermos coronarios presentan hipercolesterolemias(26). La ateroesclerosis representa en realidad una compleja cascada multifactorial donde: La inflamación; los fenómenos de crecimiento celular, posiblemente secundarios a la inflamación; la degeneración celular (depósitos de lípidos y calcificación); y las complicaciones trombóticas , representan fases del proceso, y donde su control integral va más allá de regular las cifras del colesterol plasmático (27) (Figura l3).
Bibliografia 1. Chapman MJ: Animal lipoproteins: chemistry, structure, and comparative aspects. J'Lipid Res. 1980;21:789-799.2. Green PHR, van Stiphoot Wahj, Krauss XH,et al.: Intestinal lipoprotein metabolism. J Lipid Res. 1980;21:942948. 3. Ginsberg HN.:Lipoprotein metabolism and its relation to atherosclerosis. Medical Clinics of North America. 1994;78:1-20. 4. Norun KrBerg THelgerud PDrevon CA.: Transport of I cholesterol.Physiol Rev. 1983;63:1343-1356. 5. Zilversmit DB.:Atherogenesis: A postprandial phenomenon. Circulation. 1979;60:473-485. 6. GamboaR.:1998;Observacionesnopublicadas.7. Groot PHE.:Postprandial lipoprotein metabolism in normolipidemic men with and without coronary artery disease. ArterioscierThrom.1991;1 1:653-659. 8. Patsch JP, Miesenbock G,Hopferwieser T et al.:The relation ship of triglyceride metabolism and coronary artery disease. Studies in the postprandial state. Arterioscler Throm 1992; 12:1336-1345. 9. Havel RJ.: Origin of HDL. Elsevier/North Holland Biomedical Press, Amsterdam, 1978.10.113rown MS,Goldstein JL.: The LDL receptor concept: Clinical and therapeutic implications. Atheroscler Rev.1988;18:85-89. 11. Simons LA.: Interrelaciones of lipids and lipoproteins with coronary artery disease in 19 countries. Am J Cardiol 1986;57:5G-IOG. 12. Marmot MG,Syme SL,Kagan A,et al: Epiderniologic studies ofcoronary heart disease and stroke in Japanese men living in Japan, Hawaii, and California. Am I Epidemiol 1975;102:514-18. 13. Scandinavian Simvastatin Survival Study Group. Randomized trial of cholesterol lowering in 4444 patients with coronary heart disease: the 4S Study. Lancet. 1994;344:1383-89. 14. Sacks FM,Pfeffer MA,Moye LA, et al.: The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels.N Engl J Med. 1996;335:1001-1009. 15. Shepherd J,Cobbe SM,Ford 1, et al.: Prevention of Coronary Disease with Pravastatin in men with Hypercholesterolemia. The West of Scotland Coronary Prevention Study Group.NEngIJMed. 1995;333:1301-7. 16. The LIPID Study Group: (Long Tenn Intervention with Pravastatin in Ischaemic Disease. Am J Cardin]. 1995;76:474-79. 17. Clearrield M,Stein EA,Downs JR, et al.: The Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS). Circulation. 1998;98:Supp 17,146(224). 18.Berliner JA, Heinecke JW.: The role of oxidized lipoproteins in atherogenesis. Free Rad Biol Med. 1996;20:707-727. 19. Steinberg D.:Lipoproteins and atheroesclerosis. A look back and look ahead. Arteriosclerosis. 1983;3:283-290. 20. Nordestgaard BG,and Nielsen LB.: Atherosclerosis and arterial influx of lipoproteins. Curr Opinion Lipidology.1944;5:252-257. 21. Austin MA,King MC,Vranizan KM et al.: A proposed genetic market for coronary heart disease. Circulation 1990;82:495-506. 22. Hirsch EFWeinhouse S.. The role of lipids in atherosclerosis. Physiol Rev. 194 1;23:185-190. 23. Austin MA,Goro Y, Lenfant C, et al.:The hypertriglyceridernias :risk and management. 5.Epidemiology. Am J Cardiol 1991;24:68 (3):22A-25A. 24. Assman G,Schulte H.: Procam Study.,in Lipid Metabolism Disorders and Coronary Heart Dis.2nd ed.Munich;Medizin Verlag 1993. 25. Zeiher AM,Schachinger V, Hohnloser SH.et al.: Coronary Atherosclerotic Wall Thickening and Vascular Reactivity in Humans. Circulation. 1994;89:2525-32. 26. Braunwald E. Shattuck Lecture. Cardiovascular medicine at the turn of the millennium:triumphs,concerns,and opportunities.N Engl J Med. 1997;337:1360-1369. 27.Ross R.: Atherosclerosis-An Inflarnatory Disease. N Engl J Med. 1999;340:115-126.
(*)Profesor Principal de Fisiología y Medicina Universidad Peruana Cayetano Heredia. (**)Cardiólogo Hospital Central de Policía. |