|
Genética
molecular y la fisiopatología de las enfermedades hereditarias
Dr. Ricardo Fujita Alarcón
Ph. D. (*)
Resumen
El ADN, la molécula de
la información genética tiene 2 funciones fundamentales: i) replicarse
y transmitirse sin errores, sea desde el cigoto hasta las 1014 células
de un individuo adulto, o para transmitirse a través de las generaciones;
y ii) contener en su secuencia de bases la información de las decenas
de miles de genes que codifican todas las proteínas de nuestro organismo.
Los avances de la biología molecular han permitido determinar las mutaciones
(errores) en genes que producen proteínas anómalas, o que son expresadas
en cantidades anormales en diversas patologías genéticas. En este manuscrito
se presentan algunos ejemplos de estas enfermedades y se esboza el impacto
de las mutaciones a diferentes niveles para explicar sus fenotipos.
Summary
The DNA of molecule has 2 main roles:
i) to replicate and transmit itself virtually error-free from the zygote
up to a whole adult individual, and through successive generations; and
ii) to carry the information for the tens of thousands genes coding for
all the proteins of our organism. Advances in Molecular Biology have allowed
to isolate and determine mutations and abnormal proteins which are the
primary cause of several genetic diseases. A few examples of genetic diseases
where mutations at different levels explain the phenotype shown by the
disease are presented.
"Así como
el conocimiento y la práctica actual de la medicina se basa en el conocimiento
sofisticado de la anatomía, fisiología y bioquímica, igualmente tratar
una enfermedad en el futuro requerirá un conocimiento detallado de la
anatomía molecular, fisiología y bioquímica del genoma humano…debemos
tener médicos tan buenos en anatomía y fisiología molecular de cromosomas
y genes, como el cardiólogo lo es con la estructura del corazón"(Paul
Berg, Premio Nobel de Química 1980, co-creador de la técnica del ADN recombinante).
Indudablemente
la genética molecular es ya un elemento básico en casi todas las especialidades
de la medicina contemporánea. Su importancia se refleja en la rapidez
de los avances y la generación de la información a ritmo casi exponencial,
lo que hace difícil una compilación actualizada. Por otro lado se está
creando gran expectativa debido a su potencial para resolver problemas
de salud mediante una farmacología diseńada con conocimiento de los mecanismos
moleculares de las enfermedades y la promesa de la terapia génica. En
julio de este ańo se presentó el llamado "borrador del genoma" que contenía
la secuencia de cerca del 98% de la secuencia completa de nuestro ADN
que se piensa estará completo para el 2,003. Y pensar que hasta hace menos
de 2 décadas, la genética era relegada frecuentemente al ejercicio teórico
de observación de síntomas de enfermedades genéticas. El análisis cromosómico,
algunos estudios bioquímicos y la inmunología estaban entre las pocas
ventanas para estudiar materialmente las enfermedades genéticas. Sin embargo,
estas tecnologías nunca llegaron al problema medular: caracterizar los
genes, entes que eran puramente conceptuales.
Genética Molecular
La biología molecular
"resucitó" a la genética materializando al gen, permitiendo su aislamiento,
clonación, análisis, caracterización e incluso su manipulación. Además
a partir de la secuencia de bases de un gen, se puede deducir la naturaleza
de su producto: la proteína. La disciplina híbrida de genética y biología
molecular es llamada genética molecular, que en conjunción con las ciencias
fisiológicas estudia la estructura, expresión y función de los genes y
proteínas dentro de los organismos. Gran parte del interés inicial de
la genética molecular se centró en el origen de las patologías hereditarias
monogénicas (en general raras). La búsqueda de estos genes inspiró el
llamado "Proyecto Genoma Humano" y está ampliando el interés también a
enfermedades comunes (frecuentemente mutigénicas y multifactoriales).
Actualmente se conoce genes involucrados en la propensión a diabetes,
asma, cáncer, enfermedades cardiovasculares, siquiátricas, infecciones,
suceptibilidad a fármacos o afinidad por la música entre otras características
hereditarias.
El conocimiento
de la estructura de los genes y sus proteínas han permitido hacer una
"disección molecular" de las enfermedades correlacionando los síntomas
con el tipo y lugar de mutaciones, revelando la importancia y función
de las diferentes regiones. A veces distintas mutaciones en el mismo gen
causan enfermedades clínicamente diferentes, como las distrofias musculares
de Duchenne (muy severa y letal) y la de Becker (a veces imperceptible)
causadas por mutaciones en distintas partes del gen de la distrofina.
Otro ejemplo de un gen con mutaciones con diferentes fenotipos es el gen
PPARG cuyas diferentes mutaciones están asociadas con obesidad, diabetes
tipo 2 o con cáncer al colon (ver tabla 3). La utilización
de organismos modelos mutantes, transgénicos o con genes inactivados (noqueo
de genes), así como la inmunocitoquímica permiten estudiar con más detalle
la distribución y función de la proteína normal y mutante. El análisis
de las mutaciones en humanos sirve para el diagnóstico, pronóstico y en
algunos casos prevención de enfermedades. Además el conocimiento de una
anomalía a nivel molecular permite el diseńo de terapias usando "farmacología
inteligente o dirigida" tomando en cuenta las distorsiones de proteínas
mutantes; proteínas recombinantes y la promisoria utilización en la terapia
génica.
Mutación del
ADN: Base molecular de las enfermedades hereditarias
Una carta al
editor, que sólo ocupó una página de la revista Nature, es el artículo
más trascendente en las ciencias biológicas y médicas del siglo XX (1).
Allí se revelaba la estructura del ácido desorribonucleico (ADN) formada
por las dos cadenas complementarias antiparalelas explicando 2 requisitos
para ser la molécula de la herencia: 1) que se autorreplique y pase de
célula a célula y de generación a generación y 2) que contenga información
que pueda ser traducida en caracteres.
La replicación
del ADN se basa en la complementariedad antiparalela de sus bases. Conociendo
la secuencia de una de las cadenas podemos copiar una complementaria (Ver
J. Espinoza en este mismo número). Este proceso ocurre normalmente sin
errores en la duplicación del material desde el cigoto hasta las 1014
células de un adulto. La información genética está contenida en la secuencia
de sus bases y puede ser de 2 tipos: reguladora o codante. Las secuencias
reguladoras pueden activar, silenciar, aumentar, disminuir o modular la
expresión de un gen en tejidos específicos. Las secuencias codantes contienen
una información codificada en grupo de tres bases (codones) y que es traducida
en los aminoácidos que componen las proteínas. Así las secuencias reguladoras
nos dicen cuándo, cómo y dónde se expresa un gen y las secuencias codantes
nos indican qué proteína es producida.
Eventualmente
un cambio (mutación) en la secuencia del ADN puede destruir una seńal
de regulación, cambiar un aminoácido por otro produciendo una distorsión
o truncar una proteína. Estos cambios en la expresión y estructura de
la proteína serán generalmente contraproducentes y sólo en rarísimas ocasiones
una mutación mejorará la eficiencia del producto. Cada proteína tiene
un rol bien definido en la célula y en el organismo (estructural, enzimático,
regulador, transportador, etc.), rol optimizado, en algunos casos, a través
de miles de millones de ańos de evolución. Si la mutación se produce en
células germinales (precursoras de gametos) la información de las proteínas
anómalas será transmitidas a través de generaciones.
Una proteína
anormal va a producir un disturbio en su entorno ya sea a nivel estructural
o funcional que afectará la actividad de un tejido, un órgano o un sistema
provocando frecuentemente un cuadro clínico. La severidad dependerá de
la distorsión de la proteína, la importancia del tejido, órgano o sistema
afectado; o de alternativas para compensar la anomalía (ejm. algunas isoenzimas
o vías metabólicas).
Búsqueda de
genes responsables de enfermedades
A la fecha se
han catalogado alrededor de 5,000 enfermedades genéticas, es decir anomalías
causadas por mutaciones en genes y frecuentemente heredadas. OMIM - On
Line Mendelian Inheritance in Man -, la biblia de los genetistas clínicos,
es una recopilación de enfermedades genéticas clasificadas durante muchos
ańos. OMIM es mantenida al día por Victor McKusick y su equipo y se puede
encontrar en http://www.ncbi.nlm.nih.gov/Omim/searchomim.html. Se percibe
un déficit aparente de la cantidad de enfermedades con la cantidad de
genes en el genoma (probablemente entre 40,000-80,000) y esto tiene 2
razones principales: la primera es que casi todos de genes se expresan
individualmente en estado embrionario y fetal como en un concierto sinfónico:
en el momento y con la intensidad precisa para formar, modelar y disolver
los diferentes tejidos y órganos a través del desarrollo. Una mutación
en esta mayoría de genes dará embriones y fetos con problemas para proseguir
en el desarrollo y que no llegan a término. La segunda razón es un problema
de percepción médica: lo que parece ser una enfermedad homogénea (el mismo
cuadro clínico-fenotipo), se trata en realidad de muchas enfermedades
causadas por mutaciones en genes totalmente diferentes. Tenemos el caso
de la retinitis pigmentosa (retinosis pigmentaria), degeneración progresiva
de la retina que produce visión en túnel y puede llevar hasta la ceguera
total. Mutaciones en cualquiera de los más de 30 genes localizados en
diferentes cromosomas causan la enfermedad y se cree que hay varias decenas
más por descubrir.
La citogenética
nos permite reconocer los cromosomas con una zonificación longitudinal
clara y oscura, formando las llamadas bandas cromosómicas (como ejemplo
ver figura 1). Estas bandas son constantes y características
de cada cromosoma, estimándose alrededor de 300 en todo el genoma nuclear
(22 autosomas y los cromosomas sexuales). Cada banda contiene genes específicos
(un estimado promedio de 200 genes/banda) y centenas de marcadores genéticos,
las bandas sirven de hitos en la localización y cartografía de genes.
A veces un gen no ha sido identificado; pero su localización en una banda
cromosómica es suficiente para trazar su cosegregación en una familia
con marcadores genéticos a modo de etiquetas que identifican específicamente
una región del genoma. Dentro del llamado "Proyecto del Genoma Humano"
se están localizando e identificando genes conocidos y nuevos a paso acelerado
y para el futuro inmediato queda la tarea de determinar su actividad y
averiguar su responsabilidad en enfermedades (ver Espinoza en este número).
Algunos genes
responsables de enfermedades genéticas
El estudio de
genes y proteínas involucrados en diferentes enfermedades genéticas ayuda
a entender y formular hipótesis acerca de los mecanismos por los que mutaciones
en los genes resultan en una enfermedad. A continuación se presenta una
revisión arbitraria pero tratando de mostrar diferentes enfermedades cuyos
genes responsables ya han sido identificados y que nos ayudan a explicar
la mecánica de las anomalías. Por convención, las iniciales en nomenclatura
genética son abreviaturas en inglés y se utilizará de esta manera en este
artículo.
Síndrome de Down
El síndrome de Down es un desorden genético bastante frecuente con una
frecuencia aproximada de 1: 150 embarazos y 1: 600 nacidos. El síndrome
de Down es también la causa más común de retardo mental, y ocurre con
similar frecuencia en todos los grupos étnicos. El retardo mental (de
grado variable) y la hipotonía neonatal están en el 100% de los pacientes;
hacia los treinta ańos los pacientes presentan placas amiloides en el
cerebro y con amasijos neurofibrilares, similar a la patología de Alzheimer.
Rasgos físicos conspicuos son braquicefalia, nariz achatada con puentes
elevados, epicanto, protusión de la lengua, cuello corto. Hay otros rasgos
que no son conspicuos; pero son mucho más frecuentes que en sujetos normales:
deficiencia cardiovascular, deterioro de huesos y coyunturas, inmunodeficiencia,
leucemia, cataratas, etc.
La mayoría de pacientes con síndrome de Down presenta 3 copias completas
del cromosoma 21 (trisomía libre). Sin embargo trisomías parciales con
o sin síndrome de Down delimitan a 21q22 como la zona crítica que contiene
los genes que producen el síndrome, aunque todavía no se descarta la acción
de otros genes en 21q21. La detección se hace mediante la observación
microscópica de preparaciones cromosómicas debiendo recurrir al bandeo
o a la técnica de hibridación in situ con fluorescencia (FISH) en caso
de rearreglos que involucran trisomías parciales. Cabe comentar que el
cromosoma 21 es el más pequeńo del genoma y el más pobre en genes después
del cromosoma Y, lo que explica en parte la alta viabilidad de la trisomía
21 en comparación con trisomías de otros cromosomas. Hay genes que están
óptimamente dosificados con 2 cromosomas; una copia extra derivará en
la sobreproducción de ciertas proteínas originando un desequilibrio en
la homeostasis de las células.
La relativa pobreza en genes del cromosoma 21 ha sido confirmada recientemente,
por su comparación con la secuencia del cromosoma 22, similar en tamańo
(2). Varios de los genes de la región crítica 21q21-q22 ya han sido aislados,
clonados y analizados en enfermedades humanas y modelos animales. Por
su actividad son candidatos para formar parte del grupo de genes responsables
de algunas características fenotípicas del síndrome de Down (3, 4, 5).
1. APP (proteína
precursora de placa amiloide), localizada en 21q21 cuya mutación produce
un tipo hereditario de la enfermedad de Alzheimer.
2. DSCR1 (Down syndrome critical region protein 1) es una proteína cuyo
gen está en 21q22 y se expresa exclusivamente en cerebro y corazón sugiriendo
una influencia en retardo mental y propensión a enfermedades cardiovasculares
.
3. T1AMP1 (gen del mieloma del linfocito T1) produce leucemia, que estaría
ligado a la alta incidencia en el síndrome de Down. Leucemias tipo ALL
and AML son 10-20 veces más comunes y leucemia megacariocítica aguda llega
a 200 veces la frecuencia normal, felizmente es temporal desapareciendo
en los primeros meses de vida.
4. SOD1 (sulfóxido dismutasa 1). Gen mutante en esclerosis lateral múltiple
familiar. Ratones mutantes en SOD1 tienen lenguas engrosadas y "envejecimiento
prematuro" de articulaciones.
5. MNBH (Minibrain Drosophila). Homólogo encontrado en una cepa mutante
de mosca de fruta Drosophila melanogaster que presenta un cerebro pequeńo
y problemas en "test de aprendizaje".
6. SIM (Single-minded Drosophila homolog). Gen homólogo de Drosophila,
el mutante presenta problemas en "tests de memoria".
7. COL18A1, COL6A1, COL6A2 (colágenos), expresados en tejido óseo y conectivo;
quizás asociados con características faciales, corporales y está probada
la actividad de COL6A1 yCOL6A2 en tejido cardiaco fetal en ratón. También
se demostró una propensión a problemas cardiacos asociados a alelos de
COL6A1 en pacientes Down.
8. CRYAA (Crystallin aA). Mutaciones producen catarata familiar.
9. IFNR1, IFNR2, IFNR3 (receptores de interferón), CPA1 (b integrina o
CD18) involucrados en la respuesta inmune, que puede ser asociado a la
alta propensión de infecciones en los pacientes Down. Figura
1. Esquema de cromosoma 21 con la posición relativa de los genes APP1,
SOD1, IFNR y CRYA1. Los cromosomas se dividen en bandas que contienen
genes específicos. El brazo corto se designa p y el brazo largo q. En
el síndrome de Down (nińa a la derecha) existe generalmente 3 cromosomas
21 en vez de 2, ocasionalmente hay trisomía parcial que involucra solamente
21q21 y 21q22 indicando que ésta es la región crítica para los síntomas.
| FIGURA
1 |
 |
| Esquema de cromosoma 21 con la posición
relativa de los genes APP1, SOD1, IFNR y CRYA1. Los cromosomas se dividen
en bandas que contienen genes específicos. El brazo corto se designa p y
el brazo largo q. En el síndrome de Down (nińa a la derecha) existe generalmente
3 cromosomas 21 en vez de 2, ocasionalmente hay trisomía parcial que involucra
solamente 21q21 y 21q22 indicando que ésta es la región crítica para los
síntomas. |
La
Enfermedad de Huntington
Enfermedad neurodegenerativa, hasta hace poco conocida como "corea
de Huntington" por los movimientos involuntarios que recordaban una
coreografía. El estudio patológico muestra degeneración en los núcleos
basales con la muerte de las neuronas espinosas medias. Hay perturbación
emocional con demencia progresiva, el paciente muere aproximadamente 20
ańos después del inicio de la enfermedad. Pertenece a un grupo de enfermedades
neurológicas que presenta un curioso fenómeno llamado anticipación: con
el paso de cada generación la enfermedad se expresa a menor edad. En la
enfermedad de Huntington esta anticipación está sesgada: generalmente
aparece cuando la herencia es paterna, probablemente asociada a la fisiología
de la espermatogénesis.
La enfermedad de Huntington es autosómica dominante y su gen, HD o IT15
(Huntington disease o important transcript #15) se localiza en 4p16 -
brazo corto del cromosoma 4, banda 16- y su proteína producto es la huntingtina
(6). El gen HD se expresa en el cerebro y varios otros tipos celulares
como linfocitos, tejido muscular o en gónadas; curiosamente esta expresión
no corresponde exactamente a los órganos afectados por la enfermedad.
Estudios de inmunocitoquímica muestran que la proteína huntingtina se
localiza en neuronas especialmente en el striatum coincidiendo con las
primeras áreas afectadas. A nivel subcelular la huntingtina se encuentra
asociada frecuentemente con microtúbulos en las regiones somatodendríticas
de las neuronas y también se le detecta asociada con vesículas sinápticas.
Aunque no se conoce su función exacta, se cree que la huntingtina tiene
función de anclaje o transporte de organelas (7). Experimentos en ratones
con genes "noqueados" (inactivados) revelan que mutantes homocigotos
se pierden inmediatamente después de la gastrulación revelando su papel
en el desarrollo (8).
La única mutación en HD detectada a la fecha es la expansión de un triplete
(CAG)n que normalmente se encuentra repetido de 9 a 35 veces mientras
que en pacientes se repite entre 40 a 100 repeticiones, de 36 a 39 unidades
tiene penetrancia variable (9). Este tipo de mutaciones llamadas dinámicas,
han sido descubiertas hace sólo 10 ańos, en el síndrome del X-frágil,
otra enfermedad neurodegenerativa, 2da causa de retardo mental después
del síndrome de Down, (10). Las mutaciones dinámicas son inestables y
pueden aumentar (o disminuir) en sucesivas divisiones celulares o sucesivas
generaciones. En Huntington la longitud de las expansiones están en proporción
directa con la severidad de la enfermedad e inversa con la edad de inicio:
en sucesivas generaciones hay más unidades del triplete (CAG)n. Esto dificulta
la replicación provocando en general "patinadas" de la ADN polimerasa
expandiendo (insertando) unidades extras en el ADN original (11). La expansión
a través de varias generaciones explica su aumento progresivo en longitud
haciéndola gradualmente más severa lo que explica la anticipación. La
detección y diagnóstico en laboratorio es realizada por la técnica de
la amplificación PCR del segmento genómico conteniendo la repetición (CAG)n.
El triplete CAG corresponde a glutamina en el código genético, así una
expansión de CAG dará una cantidad de glutamina anormalmente mayor. Esta
proteína mutante es reconocida por su región poliglutamina (poliGlu) y
clivada en el citoplasma por proteólisis. El segmento que contiene poliGlu
se traslada al núcleo donde precipita formando inclusiones y posiblemente
interfiera con la maquinaria de transcripción y gatille apoptosis. La
inmunohistología muestra inclusiones de la proteína mutante en los núcleos
de los ganglios basales. Se produce lo que en genética molecular se llama
"ganancia de función" o sea una propiedad nueva, ausente en
la proteína normal: La localización nuclear, la baja solubilidad y agregación
provocada por el exceso de glutamina, y posible interferencia con la transcripción
y apoptosis (12).
| FIGURA
2 |
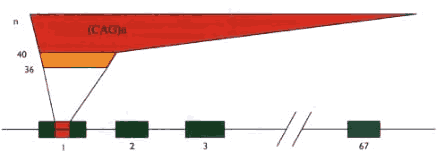 |
| Diagrama del gen HD (IT15) que tiene 67 exones y está
ubicado en el brazo corto del cromosoma 4 (4p16). Su proteína producto
(huntingtina) es rica en glutaminas. Una mutación en el exón 1 causa la
enfermedad de Huntington, la región rica en el trinucleótido (CAG) provoca
la enfermedad siempre que pasa de 40 repeticiones. Adaptado de ref. 11. |
Mucoviscidosis (Fibrosis Quística)
Enfermedad caracterizada por disfunciones en las glándulas exocrinas del
páncreas, del intestino, bronquios y glándulas sudoríparas con niveles
de cloro y sodio encima del nivel normal. La mayoría de pacientes presentan
insuficiencia de las enzimas pancreáticas con la consecuente desnutrición.
Sin embargo la mortalidad está más relacionada a problemas pulmonares,
por la formación de una mucosidad poco hidratada, viscosa, difícil de
remover por las células ciliares del tracto respiratorio. A consecuencia
de esto hay mala ventilación por estrechez de la luz bronquial, además
se acumula material contaminante facilitando la infección recurrente principalmente
por Pseudomona aeruginosa. Existe variabilidad en la afección en los diferentes
órganos y la enfermedad frecuentemente provoca la muerte antes de los
20 ańos; sin embargo hay casos menos severos con pacientes que sobreviven
hasta edades avanzadas.
La enfermedad es recesiva, su gen responsable CFTR (cystic fibrosis transmembrane
regulator) está localizado en el brazo largo del cromosoma 7 (7q21) (13).
La proteína es en sí misma un canal (o bomba) de cloro, perteneciente
a una familia de transportadores de membrana muy activos que usan la energía
del ATP. La estructura la conforman los dominios hidrofóbicos transmembranares
insertados en la capa bilipídica, 2 dominios que ligan ATP y una región
regulatoria (ver figura 3).
| FIGURA
3a |
 |
| Esquema
lineal del gen CFTR con un espectro de mutaciones y su consecuencia
en la proteína: cuadrado negro, posición de un cambio de aminionácido;
triángulo punta arriba deleción de aminoácidos; círculo azul, truncamiento;
círculo celeste, mutación de encuadre, mutación del mRNA (ref. 16). |
| FIGURA
3b |
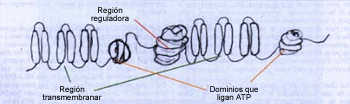 |
| Esquema de la proteína "desdoblada", mostrando
la estructura secundaria con los diferentes dominios y regiones (adaptado
de ref. 16). |
| FIGURA
3c |
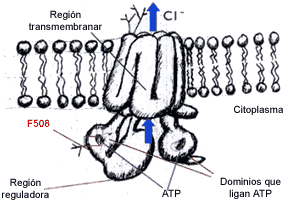 |
| Representación
terciaria y cuaternaria de la proteína CFTR con su posición en la
membrana celular y la disposición de los dominios, regiones y la región
afectada por la mutación A508F. |
Se
han detectado alrededor de 800 diferentes mutaciones correlacionadas con
diferentes fenotipos. El 70% de los pacientes (caucásicos en Europa y
Norteamérica) tienen una mutación (llamada DF508) que elimina la fenilalanina
del codón (aminoácido) 508. Esta fenilalanina es el sitio de una seńal
de glicosilación -como tarjeta de embarque- para trasladar CFTR desde
el retículo endoplásmico a la membrana celular (a través del Golgi). La
proteína mutante es rápidamente reconocida por un sistema que degrada
proteínas de membrana/secreción defectuosas y es eliminada en el mismo
retículo, por lo tanto no se insertará en la membrana como normalmente
corresponde (14).
Una mutación que inactiva la proteína funcional como (DF508), que trunque,
no produzca o distorsione drásticamente la proteína, provocará una mucoviscidosis
más severa con insuficiencia pancreática además del problema bronquial.
Un ejemplo es la mutación Gln551Asp que cambia una glicina (básica) por
un aspartato (ácido) en una zona de adhesión del ATP, lo que inactiva
el transporte de cloro a pesar de ubicarse correctamente en la membrana
(14, 15) . Una mutación que no cambie drásticamente la naturaleza del
aminoácido dará una proteína subóptima; pero parcialmente funcional. La
enfermedad en estos casos es menos severa, usualmente con actividad normal
o aceptable del páncreas, como el caso de la mutación Arg117His que cambia
una arginina (aminoácido básico) por una histidina (también básico) (15).
Experimentos in vitro han demostrado que sólo se necesita alrededor de
6% de células expresando un gen CFTR normal para que la población tisular
tenga una actividad exocrina normal. Esto explica la recesividad de esta
enfermedad: una persona heterocigota (un gen afectado y uno normal) produce
más que suficiente (50%) para suplir la necesidad de esta proteína en
los tejidos. Esta razón será también la base de estrategias para la corrección
mediante terapia génica; concentrándose quizás en una reducida población
de las células del tracto respiratorio, ya que estas determinan la vida
de los pacientes (16).
Diabetes mellitus
En los números 1 y 2 del volumen 39 de esta revista se presentó un simposio
muy completo sobre diabetes mellitus revisándose los conceptos clínicos
y las complicaciones en los diferentes sistemas. Aunque el aporte genético
se menciona directamente en un artículo (17), en los otros se puede vislumbrar
la participación de distintos genes. Esto es sugerido por la variedad
de síntomas, las diferentes proteínas involucradas y las variaciones poblacionales.
La diabetes es uno de los paradigmas de las enfermedades multifactoriales
o complejas, producidas por la conjunción factores ambientales y genes
con influencia variable. Una mutación en un gen predispone; pero no siempre
es factor suficiente para provocar la enfermedad. Esto contrasta con la
mayoría de las enfermedades monogénicas (mendelianas), como Huntington
o mucoviscidosis donde la mutación en un gen es suficiente para desencadenar
la patología. En enfermedades complejas el riesgo de tener la enfermedad
es del orden de 5-15% en cada generación, se dice que la enfermedad "corre
en familias" aunque en pocas familias parezca herencia mendeliana
(con riesgo de 25 a 50%). Esta herencia atípica hizo que la diabetes fuera
vista hasta hace poco una pesadilla para los genetistas.
| TABLA
1 |
| CLASIFICACIÓN
CLINICA de DIABETES (tomado de ref. 18) |
| |
Tipo
I (IDDM) |
Tipo
2 (NIDDM) |
MODY |
|
Edad
inicial
Proporción
Control
de hiperglicemia
Riesgo
en hermano
Obesidad
|
Juvenil
0.4%
(Reino Unido)
Requiere
insulina
6-10%
No
|
>
40 años
6%
(EEUU)
Hipoglucemia
oral
10-15%
Asociación
fuerte
|
Juvenil
Rara
Hipoglucemia
oral
Autosómico dominante (50%)
No
|
La clasificación de la diabetes desde el punto de vista genético ha sido
facilitada por la distinción clínica de 2 tipos principales de diabetes:
tipo 1 ó IDDM (insuline-dependent diabetes mellitus, diabetes tipo 2 ó
NIDDM (non-insuline-dependent diabetes mellitus). En la IDDM hay carencia
de insulina, aparece a corta edad y está asociada con obesidad. En la
NIDDM hay insulina; pero las células blanco no responden al estímulo,
el inicio de la enfermedad es generalmente en la edad madura, se puede
controlar con dieta y está frecuentemente asociada con obesidad. Además
hay una tercera variante llamada MODY (maturity-onset diabetes of the
young), poco frecuente, de herencia dominante, no dependiente de insulina;
pero la edad de inicio en la nińez o pubertad. El análisis molecular ha
mostrado que mutaciones en un mismo gen pueden causar MODY o NIDDM dependiendo
de la naturaleza de la mutación (ver Tabla 3).
Los avances con el estudio de genes y del genoma han permitido refinar
los estudios y demostrar que la diabetes es en realidad una constelación
de enfermedades provocadas en parte por mutaciones en decenas de genes
y que convergen en síntomas similares.
| TABLA
2 |
GENES
PARCIALMENTE RESPONSABLES de la DIABETES MELLITUS INSULINO DEPENDIENTE
(IDDM o diabtes tipo i).
El
locus indica que se sabe que un gen reside en una región del
genoma, que puede o no estar caracterizado. |
| Locus |
Localización |
Proteína/Gen
conocidos |
IDDM1
IDDM2
IDDM3
IDDM4
IDDM5
IDDM6
IDDM7
IDDM8
IDDM11
IDDM12
IDDM13
IDDM15
IDDM17
|
6p21
11p15
15q26
11q13
6q24-q27
18q21
2q31
6q25-q27
14q24-q31
2q33
2q34
6p21
10q25
|
DQB/DQB
del HLa (MHC)
Insulina / INS
desconocido
desconocido
desconocido
desconocido
NEURODI / (neuronal
development) a confirmar
desconocido
desconocido
CTLA4 (cytotoxic Tlymphocyte associated 4)
desconocido
desconocido (No HLA)
Detectado en familias de
beduinos árabes |
La IDDM es producida principalmente por autoinmunidad a la insulina o
a proteínas de los islotes de Langerhans y el gen más frecuente fue denominado
IDDM1. Estudios iniciales indicaban como candidato IDDM1 al gen HLA-DR
(con los alelos DR3 y DR4) del cluster HLA (Human Leukocyte Antigen),
parte del complejo mayor de histocompatibilidad en 6p21. Sin embargo estudios
posteriores seńalan al vecino gen HLA-DQb (proteína DQB) que presenta
una mayor correlación entre mutaciones y la enfermedad. Un aspartato en
posición 57 (asp57) en DQB parece importante para un funcionamiento normal:
96% de los pacientes IDDM1 carece de asp57 en ambos cromosomas, presentando
otro aminoácido en esa posición, mientras que esto se observa sólo en
19% de personas normales (19). Estudios cristalográficos muestran que
DQB tiene una conformación distinta a los otros HLA en los alrededores
de asp57, que le da preferencia por péptidos específicos, un cambio de
aminoácido distorsionaría este sitio de reconocimiento (20). Otro gen
en IDDM es por supuesto, el gen de la insulina (INS referida a la proteína,
IMMD2 referida a la enfermedad), localizado en 11p15. Hay indicación de
otros genes que han sido mapeados a distintas regiones del genoma; pero
aún no han sido aislados y caracterizados como IDDM4 en 11q13, IDDM5 en
6q25, IDDM8 en 6q27 y IDDM12 en 2q33.
Mientras que algunos genes propuestos como IMMD han sido descartados como
artefactos del análisis estadístico, como por ejemplo IDMM5 (21).
Los análisis genéticos en NIDDM son menos claros que los IDDM, parece
haber más genes; pero con menos influencia en la enfermedad. Como consecuencia,
la variación genética en NIDDM es más marcada y los genes parecen tener
diferente prevalencia en las distintas poblaciones estudiadas. En algunos
grupos nativos de Norteamérica, Sudáfrica y Finlandia hay asociación con
alelos del grupo HLA lo que indicaría que uno de los genes de la región
estaría involucrado en la enfermedad en estas etnias. En los descendientes
de mejicanos en EE.UU. hay genes que están localizados en 2q37, 16q22
y 19p13.
| TABLA
3 |
ALGUNOS
LOCI-GENES ASOCIADOS a MODY y a la PROPENSIÓN de IDDM (diabetes
tipo I) y NIDDM (diabetes tipo 2). Dependiendo de la naturaleza y
la posición de una mutación en el mismo gen, aquella
puede causar distintas enfermedades.
Ver GCK, PPARG o DCP/ACE |
| Locus |
Localización |
Proteína/Gen
conocidos |
MODY
1
MODY2-NIDDM-
Diabetes mellitus gestacional
MODY3-IDDM
MODY4-IDDM
MODY5
NIDDM1
NIDDM2
NIDDM3
NEURODI
PPARG
Dipeptidil
carboxipeptidasa I |
20q12-q13
7p15-p13
12q24.2
13q12.1
17cen-q21.3
2q
12q
20q
2q31
3p25
17q23 |
Factor
nuclear del hepatocito 4 alfa (HNF4A)
Glucoquinasa (GCK) / Hexoquinasa 4 /HK4)
Factor nuclear del hepatocito 1 alfa (HNF1A)
Factor promotor de insulina 1 (IPFI)
Factor de transcripción 2 (TCF2)
Mejico-amercianos de Texas / desconocido
Desconocido, diferente a HNF1A
Desconocido, diferente a HNF4A
NEURODI / (neuronal development)
PPARG (Receptor y del peroxisoma proliferador-activado).
Diferentes mutaciones asociadas con: obesidad, cáncer al colon
(mutaciones somáticas).
NIDDM con acanthosis nigricans e hipertensión.
DCPI / ACE (angiotensin converting enzyme).
Suceptibilidad a nefropatía diabética y enfermedades
cardiovasculares. |
Trabajos clásicos con los nativos del Suroeste de EE.UU., especialmente
la etnia Pima de Nuevo Méjico, muestran alta incidencia de NIMMD y obesidad
en condiciones de vida occidentalizada (sedentaria, con dieta con exceso
de azúcar y grasa). La contraparte son los miembros de la misma etnia
que se separaron hace algunos cientos de ańos y que viven en México en
condiciones de vida no occidentalizadas y que no presentan problemas de
diabetes ni obesidad: las 2 poblaciones son casi idénticas genéticamente;
pero el estilo de vida desencadena la anomalía. Al menos 4 genes de suceptibilidad
pre-diabética en el grupo Pima han sido mapeados tentativamente en 3q21-24
(concentración de insulina en ayunas y acción in vivo), 4p15-q12 (concentration
de insulina en ayunas), 9q21 (test de tolerancia a glucosa oral, concentración
de insulina luego de 2 horas) y 22q12-13 (glucosa plasmática en ayunas)
(22).
Estudios de segregación en una gran familia con MODY sirvieron para apuntar
al gen de la glucoquinasa (GCK ó MODY2 en 7p15-p13), que mostró ulteriormente
mutaciones en más familias con MODY y otras mutaciones con familias con
NIDDM. A la fecha se han determinado 5 genes que producen MODY con ciertas
mutaciones y NIDDM con otras. Los genetistas asignan los genes de acuerdo
a la enfermedad y un número, este nombre puede tener sinónimos cuando
el gen y su proteína han sido identificadas, como el ejemplo MODY2 = gen
de la glucoquinasa (GCK).
Por otro lado los modelos animales pueden dar luces de procesos que todavía
no detectamos en anomalías humanas: hay un alelo (variante genética) del
gen IRS-1 (insulin receptor substrate # 1) que es bastante más frecuente
en los pacientes con NIDDM que en la población normal; pero se ignora
su rol en la enfermedad. La proteína IRS-1 es sustrato de la tirosin kinasa
del receptor de la insulina (INSR), y probablemente tenga función de adherir
y activar otras seńales de transducción luego de ser fosforilada. La deficiencia
de IRS-1 en ratones produce una ligera hiperglicemia apenas detectable.
Una proteína análoga llamada IRS-2 fue identificada en ratones produciéndoles
NIMMD cuando se inactiva, e igualmente se desconoce su rol en humanos.
Para analizar el efecto aditivo de estos genes se inactivaron IRS-1, IRS-2
y INSR en ratones y luego se cruzaron líneas para obtener combinaciones
de los distintos genes en heterocigosis. La diabetes se desarrolló en
40% de los ratones con la inactivación heterocigota de los 3 genes a la
vez, en 20% para INRS e IRS-1, en 17% para INRS e IRS-2 y en 5% para INRS
solo. A pesar que las combinaciones INSR IRS-1 e INRS IRS-2 dan número
parecidos, la fisiología era diferente. Mutantes INSR IRS-1 produce resistencia
severa a la insulina en músculo esquelético e hígado con hiperplasia de
células b. Mientras que mutantes INSR IRS-2 producía resistencia severa
en hígado, resistencia leve en músculo esquelético e hiperplasia modesta
de células b (23).
Conclusiones y Perspectivas
Las enfermedades hereditarias son causadas por falta, deficiencia o distorsión
de proteínas que a su vez han sido provocadas por errores en la información
genética contenida en la secuencia de ADN. La genética molecular nacida
de la conjunción de la genética y las técnicas de biología molecular ha
permitido analizar directamente los genes y proteínas que están involucradas
en enfermedades hereditarias.
La mayoría de las enfermedades hereditarias carecen de causa primaria
conocida, es decir no se ha caracterizado el gen/la proteína deficiente
que las produce, aunque a la fecha decenas de genes y proteínas han sido
identificados como causa o predisposición de algunas de ellas. Ejemplos
como los genes cuyas mutaciones provocan la mucoviscidosis (CFTR), la
enfermedad de Huntington (HD) u otros genes que predisponen a la diabetes
(INS, DQB, GCK). La comparación entre los genes normales y mutados dan
una explicación del mecanismo causal en estas enfermedades. Por otro lado
los avances en el Proyecto del Genoma Humano están revelando decenas de
miles de genes localizados en todas las regiones cromosómicas. Aunque
la mayor parte de estos genes son desconocidos, el próximo paso será estudiar
sus funciones. Los genes de enfermedades localizadas en regiones cromosómicas
específicas (por ejemplo varios de los IDDM o NIDDM) contarán en adelante
con un catálogo de genes candidatos de la región para verificar la presencia
de mutaciones en pacientes.
El análisis del gen y su proteína nos permite conocer las causas íntimas
de las enfermedades genéticas, sean estas provocadas por la mutación en
un sólo gen o por la interacción de varios genes mutados. Ahora podemos
explicar a nivel molecular y celular la razón de algunas enfermedades
recesivas o dominantes ya que la lesión genética ha sido determinada,
la anomalía en su proteína comprobada y en muchos casos se ha podido correlacionar
el tipo mutación con la severidad de fenotipos. Los mecanismos de las
mutaciones y la mecánica molecular y celular de las patologías son variados,
aquí se ha presentado algunos ejemplos; tratando de demostrar la causalidad
de las mutaciones en la presentación del fenotipo.
Además conocer el gen será útil para diagnóstico y pronóstico de enfermedades
determinando quién tiene el gen afectado en una familia. Por otro lado
este conocimiento sirve para la búsqueda de terapias farmacológicas "inteligentes"
o dirigidas en base al conocimiento de la proteína; modificación o reemplazo
de la proteína defectuosa/ausente con terapia génica.
Las perspectivas para el estudio de la genética molecular en enfermedades
en nuestro país son grandes; pero poco se ha hecho en el campo y los trabajos
con muestras peruanas han sido generalmente procesados en el exterior.
En el Perú tenemos una gran variedad de grupos étnicos nativos y otros
que migraron después de la conquista, con un bagaje genético propio de
enfermedades y de frecuencias que resta a explorar y será necesario estudiar.
De especial importancia en salud pública será determinar la propensión
genética a enfermedades comunes como las cardiovasculares, diabetes, asma,
cáncer o incluso determinar la propensión a las infecciones o sensibilidad
a los fármacos. Si bien en corto o mediano plazo no contaremos con la
terapia génica en estas enfermedades, si es posible identificar las personas
con mutaciones para hacerles un seguimiento cuidadoso y monitorear los
factores externos como dietas, ejercicios, etc.
Bibliografía
1. Watson, J.D. and Crick, F.H.C. A structure for deoxyribose nucleic
acid. Nature 1953; 171: 737.
2. Hattori, M;, Fujiyama, A; Taylor, T.D; Watanabe, H. et. al. The
DNA sequence of human chromosome 21. Nature 2000; 405: 311-319.
3. Hernandez, D. and Fisher, E.M.C. Down syndrome genetics: unravelling
a multifactorial disorder. Hum. Mol. Genet. 1996; 5: 1411-1416.
4. Kola, I; Hertzog, P.J. Animal model in the study of the biological
function of genes on human chromosome 21 and their role in the pathophysiology
of Down syndrome. Hum. Mol. Genet. 1997; 6: 1713-1727.
5. Smith, D.J. and Rubin, E.M. Functional screening and complex
traits: human 21q22.2 sequences affecting learning in mice. Hum. Mol.
Genet. 1997; 6: 1729-1733.
6. Huntington's Disease Collaborative Research Group. A novel gene
containing a trinucleotide repeat that is expanded and unstable on Huntington's
disease chromosomes. Cell 1993; 72: 971-983.
7. Gutekunst, C.-A; Levey, A. I; Heilman, C. J; Whaley, W. L. et al.
Identification and localization of huntingtin in brain and human lymphoblastoid
cell lines with anti-fusion protein antibodies. Proc. Nat. Acad. Sci.
1995; 92: 8710-8714.
8. MacDonald, M. E; Duyao, M; Calzonetti, T; Auerbach, A. et al. Targeted
inactivation of the mouse Huntington's disease gene homolog Hdh. Cold
Spring Harbor Symp.Quant. Biol. 1996; 61: 627-638.
9. The American College of Medical Genetics/American Society of Human
Genetics, Huntington Disease Genetic Testing Working Group. ACMG/ASHG
Statement: Laboratory guidelines for Huntington disease genetic testing.
Am. J. Hum. Genet. 1998; 62:1243-1247.
10. Oberle, I; Rousseau, F; Heitz, D; Kretz, C. et al. Instability
of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome.
Science 1991; 252:1097-1102.
11. Read, A. P. Huntington's disease: testing the test. Nature
Genet. 1993; 4: 329-330.
12. Gusella, J. F; McNeil, S; Persichetti, F., Srinidhi, J. et al.
Huntington's disease. Cold Spring Harbor Symp. Quant. Biol. 1996; 61:
615-626.
13. Kerem, B; Rommens, J. M; Buchanan, J. A. Markiewicz, et al. Identification
of the cystic fibrosis gene: genetic analysis. Science 1989; 245: 1073-1080.
14. Jensen, T. J., Loo, M. A; Pind, S; Williams, D. B. et al. Multiple
proteolytic systems, including the proteasome, contribute to CFTR processing.
Cell 1995; 83: 129-135.
15. Logan, J; Hiestand, D., Daram, P., Huang, Z. et al. Cystic
fibrosis transmembrane conductance regulator mutations that disrupt nucleotide
binding. J. Clin. Invest. 1994; 94: 228-236.
16. Cox, T.M. and Sinclair, J. Molecular Biology in Medicine. Blackwell
Science Ltd. (1997).
17. Battilana, C. y Lu, L. Nefropatía diabética. Diagnóstico 2,000;
39:66-79.
18. Strachan, T. and Read, A.P. Human Molecular Genetics. Bios
Scientific Publishers Limited. (1996).
19. Todd, JA;Bell, JI.and McDevitt, HO. HLA-DQ(beta) gene contributes
to susceptibility and resistance to insulin-dependent diabetes mellitus.
Nature 1987; 329: 599-604.
20. Corper, AL; Stratmann, T; Apostolopoulos, V; Scott, CA. et al.
A structural framework for deciphering the link between I-AG7 and
autoimmune diabetes. Science 2,000; 288: 505-511.
21. Nistico, L; Buzzetti, R; Pritchard, LE; Van der Auwera, B.
et al. The CTLA-4 region of chromososme 2q33 is linked to, and associated
with, type 1 diabetes. Hum. Molec. Genet. 1996; 5: 1075-1080.
22. Pratley, RE; Thompson, DB; Prochazka, M; Baier, L. et al. An
autosomal genomic scan for loci linked to prediabetic phenotypes in Pima
Indians. J. Clin. Invest. 1998; 10: 1757-1764.
23. Kido, Y; Burks, DJ, Withers, D; Bruning, JC. et al. Tissue-specific
insulin resistance in mice with mutations in the insulin receptor, IRS-1,
and IRS-2. J. Clin. Invest. 2,000; 105: 199-205.
(*) Profesor Investigador,
Instituto de Génetica y Biología Molecular - Facultad de
Medicina Humana, Universidad de San Martín de Porres
|