|
Investigación
clínica y genética de una familia peruana afectada por Ataxia Espinocerebelosa
tipo7 (AEC7)
Dres. Marco Antonio Castańeda
(*)
Carmen Avalos (**)
Raúl Jerí (*)
Resumen
El estudio clínico y genético de las atrofias del cerebelo, enfermedades
familiares o esporádicas, ha experimentado recientemente una verdadera
revolución al describirse la localización de la enfermedad en los genes
cromosómicos. Este tipo de investigación recién se inicia en nuestro país
con una familia peruana procedente de Huarochirí. Se examinó a la familia,
que consistía en 65 personas, en cinco generaciones, afectadas con diversos
trastornos neurológicos asociados a perturbaciones del equilibrio. El
examen consistió en evaluación neurológica, neuropsicológica, oftalmológica,
neuroradiológica y genética. Esta última investigación se realizó en el
laboratorio de neurogenética de la Universidad de Utah (Salt Lake City).
Los síntomas y signos consistían en perturbaciones del equilibrio, dificultad
para caminar, debilidad de las extremidades, diversos grados de ataxia,
piramidalismo, oftalmoplejía, variable compromiso de la agudeza visual,
maculopatía, degeneración retiniana, atrofia de los nervios ópticos, atrofia
del cerebelo, del tronco encefálico y de los hemisferios cerebrales. El
ADN de 30 individuos mostró en los afectados expansión repetida de los
tripletes CAG en el brazo corto del cromosoma 3p. Los hallazgos comprobados
en la familia examinada demuestran que se trata de ataxia espinocerebelosa
tipo 7 (AEC7).
Palabras clave: Ataxia Espinocerebelosa, AEC7, Aspectos Genéticos, AEC7
en el Perú.
Summary
Recently the clinical and genetic investigation of cerebellar diseasses
have experienced an extraordinary advance due to localization of the disorders
in different chromosomal loci. These type of investigation have been done
in Peru in the last decade, studying a peruvian family from the Huarochiri
province. A peruvian family of 65 individuals in five generations, affected
by several neurological disturbances associated with ataxia, were studied
by clinical, neurological, opthalmological, neuroradiological and genetic
methods. The genetic study was done in the neurogenetics laboratory of
the University of Utah (Salt Lake City). The patients showed equilibrium
disturbances, gait difficultics, opthalmoplegia, corticospinal signs,
cognitive deficiencies, visual weakness, maculopathy, retinal degeneration,
optic nerve atrophy and atrophy of the cerebellum, brain stem and cerebral
hemispheres. The DNA in 30 individuals showed, in the neurologically affected,
repeated expansion of CAG triplets in the short arm of chromosome 3p.
The findings in this family suggests that they suffer from spinocerebellar
ataxia type 7 (AEC7).
Key Words: Spinocerebellar ataxia, AEC7, Genetics, AEC7 in Peru.
Introducción
Las ataxias espinocerebelosas autosómicas dominantes son un grupo heterogéneo
de desórdenes neurodegenerativos (1). Varias clasificaciones clínicas
y neuropatológicas han sido establecidas, basadas en la edad de inicio
y el grado de compromiso del cerebelo, los ganglios basales, la médula
espinal, el tronco encefálico y la presencia de neuropatía o retinopatía
(2, 3).
La identificación de varios y distintos genes ha permitido un esquema
de clasificación basado en el genotipo (4).
En las ataxias espinocerebelosas (AEC) 1, 2, 3, 6 y 7, las mutaciones
genéticas han sido tipificadas y comprometen una expansión repetida del
trinucleótido CAG en la región del gen codificado (5, 6). Las ataxias
espinocerebelosas tipo 4 (AEC4) se han ligado al cromosoma 16 y la AEC5
al cromosoma 11, sin embargo, al momento los genes están por identificarse
en las AEC (7- 8).
Las ataxias espinocerebelosas tipo 7 (AEC7) presentes en varias familias
han sido descritas por L. Gouw y colaboradores en donde al defecto atáxico
se asocian trastornos córticoespinales, nistagmo, oftalmoplejía y distrofia
pigmentaria macular que conduce a la ceguera (9).
La nomenclatura previa para la AEC7 incluye ataxia cerebelosa autosómica
dominante (ACDA) tipo II (10) y atrofia olivopontocerebelosa (AOPC) tipo
III (11). Los estudios de encadenamiento para muchos autores han mapeado
al gen causante de la enfermedad en el brazo corto del cromosoma 3 (3p12)
(12, 13).
En la AEC7, al igual que en el resto de las ataxias cerebelosas autosómicas
dominantes y otras producidas por la expansión del triplete CAG, se produce
anticipación genética (14)., esto es, el número de repeticiones es inversamente
proporcional a la edad de aparición de los síntomas y a la intensidad
de la afectación clínica (15).
Nosotros describimos en este trabajo la primera investigación clínica
y genética de una familia peruana afectada por ataxia espinocerebelosa
tipo 7 (AEC7).
Método
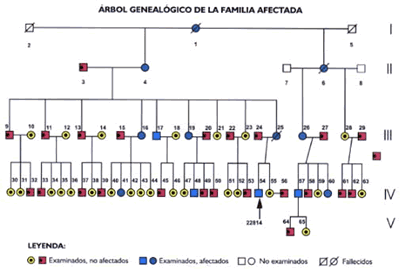
El paciente índice (IV-54) de la familia afectada (Figura
1) fue inicialmente identificado y evaluado en el Consultorio de Neurología
del Hospital Nacional Dos de Mayo por uno de los autores. Se trataba de
un varón de 23 ańos de edad con una historia de 8 ańos de enfermedad consistente
en disturbios para la marcha y defecto visual. En abril 2 de 1998 el enfermo
estaba incapacitado para mantenerse en posición de pie, postrado en cama
con severa incoordinación de los cuatro miembros, disartria, disfagia,
marcado temblor al movimiento intencional, espasticidad, hiperreflexia
osteotendinosa y signos de Babinski y Hoffman bilaterales. La evaluación
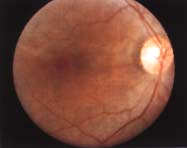
oftalmológica determinó no percepción visual de luz, severa palidez de
los nervios ópticos, atenuación de los vasos de la retina, atrofia macular,
ver (Fig. 1C, 1D), vasculitis, hemorragias y atrofia
retinal verificada por fundoscopía y estudio con angiofluoresceína. Además
se demostró ausencia de nistagmo optocinético y oftalmoplejia. Finalmente
la valoración del estado mental mediante la prueba de la Escala de la
Inteligencia para adultos (WAIS) dio como resultado 72 en el percentil
verbal (no se determinó el percentil ejecutivo por el compromiso visual).
La evaluación fue complementada mediante el Test Minimental de Folstein
con una puntuación de 23, lo que permitió concluir que existía moderado
deterioro intelectual.
| FIGURA
1 |
 |
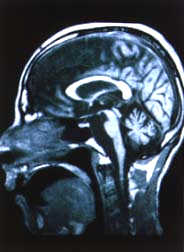
El estudio con resonancia magnética (RMN) del encéfalo mostró atrofia
pontocerebelosa y de los hemisferios cerebrales (Figs.
1A y 1B).
Después de la identificación del probando,
los miembros adicionales del árbol genealógico fueron ubicados en diferentes
lugares del Departamento de Lima, en regiones de la serranía (Distrito
de San Gerónimo de Surco, Provincia de Huarochirí, a 2780 msnm) y en zonas
marginales de la ciudad.
OBSERVACIÓN
# 1
Caso Indice: IV - 54; DNA 22814; varón de 23 años de edad;
59 CAG
| FIGURA
1A |
FIGURA
1B |
 |
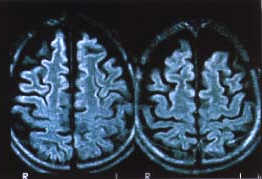 |
| Estudio
con RMN del encéfalo: Considerable atrofia del tronco cerebral,
cerebelo y los hemisferios cerebrales. |
| FIGURA
1C |
FIGURA
1D |
 |
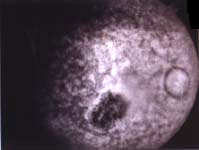 |
| Fondo
del ojo y estudio con angiofluoresceina: Atrofia de la papila óptica,
vasculitis, extensa distrofia macular, retinopatía distrófica
y microhemorragias. |
Estos pacientes fueron examinados en los aspectos neurológico, neuropsicológico,
oftalmológico (determinación de la agudeza visual, precisión de los campos
visuales mediante campimetría computarizada perimétrica, capacidad de
discriminación de colores mediante las pruebas de Ishihara, Farnsworth
y Roth) y se realizaron angiografías con fluoresceína en ocho pacientes
afectados clínicamente por ataxia cerebelosa y maculopatía distrófica
progresiva.
Se efectuaron en tres pacientes
(II-4; IV-54 y IV-57) estudios con neuroimágenes mediante tomografía axial
computarizada del cerebro (II -4) y resonancias magnéticas (IV-54 y IV-57).
Finalmente en 30 de las 63 personas, se extrajeron muestras de sangre
venosa, (20 cc por cada paciente), las cuales fueron enviadas al laboratorio
del Departamento de Neurogénetica de la Universidad de Utha-USA para el
estudio del ADN y el encadenamiento cromosómico, previa autorización de
los pacientes (Tabla 1).
Resultados
Las observaciones clínicas se efectuaron en una familia con 65 miembros
(31 varones y 34 mujeres). Trece estuvieron clínicamente afectados, diez
estaban vivos y tres habían fallecido con una historia de ceguera y ataxia
axial y de los miembros, dismetría, disdiadococinesia y disartria, piramidalismo
y en tres pacientes más comprometidos hubo disfagia. Los tests neuropsicológicos
para valorar la función cognitiva global, mediante el mini-examen cognitivo
de Folstein, las matrices progresivas de Raven y la escala de inteligencia
para adultos (WAIS), efectuados en 8 pacientes dieron por resultado rangos
variados de deterioro intelectual. Igualmente el compromiso de la motilidad
ocular y la presencia del nistagmo se comprobó en 7 pacientes, finalmente
en todos ellos hubo disminución de los movimientos sacádicos de los ojos
o ausencia del reflejo optocinético.
| TABLA
1 |
| RESULTADOS
DEL ESTUDIO DEL ADN Y EL NÚMERO DE EXPANSIONES AS CAG |
| IG |
ID.DNA |
Condición |
Edad |
Sexo |
T.
de Enferm.* |
Nº
Expans. CAG |
II-14
III-16
III-17
III-21
III-13
III-22
IV-54
IV-40
IV-39
IV-38
IV-41
III-11
III-12
III-33
IV-34
III-9
III-14
III-18
II-33
III-26
IV-57
IV-59
IV-60
III-28
IV-63
IV-62
IV-53
III-19
IV-48
IV-47
|
22808
22809
22810
22811
22812
22813
22814
22815
22816
22817
22818
22819
22820
22821
22822
22823
22825
22826
22827
22865
22866
22867
22868
22869
22870
22871
22872
22873
22875
22876
|
Afectada
Afectada
Afectado
No
No
No
Afectado
No
No
No
Afectada
No
No
No
No
No
No
No
No
Afectada
Afectado
No
Afectada
No
No
No
No
Afectada
Afectado
No
|
61a.
34a.
31a.
30a.
28a.
36a.
23a.
14a.
12a.
13a.
17a.
36a.
26a.
13a.
12a.
38a.
32a.
30a.
62a.
56a.
36a.
28a.
24a.
52a.
48a.
46a.
12a.
22a.
10a.
12 a.
|
F
F
M
F
M
M
M
M
M
F
F
M
M
M
M
M
M
M
M
F
M
F
F
F
M
F
M
F
M
M
|
13a.
10a.
8a.
-
-
-
10 a.
-
-
-
2
-
-
-
-
-
-
-
-
11a.
18a.
-
8a.
-
-
-
-
6a.
2a.
-
|
44
47
47
10
13
13
59
12
13
13
51
13
10
10
10
13
10
12
13
42
53
12
48
10
12
10
10
47
63
12
|
La mayoría de los enfermos
con definida expresión fenotípica se encontraban francamente atáxicos,
al extremo que necesitaban ayuda de la familia para deambular., tres pacientes
se encontraban postrados en cama por el severo trastorno atáxico y les
era imposible mantenerse en pie o iniciar la marcha; solamente en un nińo
de diez ańos (IV-48) el trastorno fue leve, consistente en dificultades
al correr o saltar que le habían provocado muchas caídas., sin embargo,
la marcha habitual y la prueba sensibilizada del Romberg fueron normales.
En ninguno de los pacientes se encontraron trastornos objetivos de la
sensibilidad ni signos extrapiramidales.
En tres pacientes (II-4; IV-54 y IV-57) se realizaron estudios con neuroimágenes,
mediante estudio con tomografía axial computarizada del cerebro (II-4)
y RMN (IV-54 y IV-57), los cuales revelaron grave atrofia del cerebelo,
el tronco encefálico y de los hemisferios cerebrales.
La evaluación oftalmológica, inicialmente efectuada en el servicio de
Oftalmología del Hospital Nacional Dos de Mayo, fue complementada y ampliada
en un centro oftalmológico computarizado. Las evaluaciones en trece pacientes
de la familia problema (10 afectados) consistieron en la determinación
de la agudeza visual, obteniéndose grados variables de compromiso de esta
función, encontrándose en tres enfermos severo trastorno al no percibir
la luz. De otro lado se preciso la capacidad de discriminación de colores
mediante la aplicación de los test de Ishihara, el Farnsworth D-15 y el
test de 28 matices de Roth (Modificado del Farnsworth 100 cada 3 cápsulas
de color). Los pacientes no tuvieron problemas en realizar la prueba y
ninguna de ellas indicó eje de error, algunos tuvieron errores leves que
no alteraron la circunferencia de las gráficas.
El test de colores falló en detectar casos precoces de compromiso ocular
ya sea macular o del nervio óptico.
En seis pacientes se realizaron diferentes pruebas del campo visual con
el perímetro computarizado de Humphrey. En dos enfermos, uno con agudeza
visual 20/30 y otro con 20/50 en ambos ojos, la perimetría demostró depresión
generalizada y caso dudoso, respectivamente. También se aplicaron en algunos
pacientes pruebas especiales como mácula con índice blanco y mácula con
índice rojo, tratando de ver si alguna de ellas podría ser útil en la
detección de casos precoces., el resultado fue negativo.
OBSERVACIÓN
# 2
Caso Indice: IV - 57; DNA 22866; varón de 36 años de edad;
53 CAG
| FIGURA
2A |
FIGURA
2B |
 |
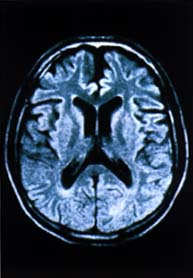 |
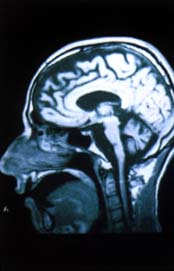
| Estudio
con RMN del encéfalo: Atrofia del tronco encefálico,
del cerebelo y los hemisferios cerebrales |
| FIGURA
2C |
FIGURA
2D |
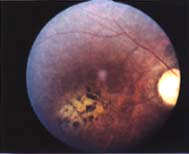 |
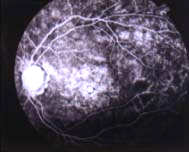 |
| Fondo
del ojo y estudio con angiofluoresceina: Atrofia del nervio óptico,
maculopatía y distrofia difusa retinal. |
Se realizaron angiogramas
con fluoresceína en ocho pacientes. En todos los pacientes con compromiso
severo y en algunos sin compromiso evidente en la oftalmoscopía, esta
técnica fue muy útil para trasladar a una gráfica la maculopatía atrófica.
La motilidad ocular estuvo comprometida en siete de los enfermos, en dos
de ellos casi en estado de oftalmoplejía (IV-54; y IV-57). En cinco casos
se encontró estado de oftalmoparesia y tres tuvieron preservación de la
motilidad ocular. Los movimientos sacádicos estuvieron abolidos en tres
enfermos y disminuidos en cuatro, tres casos con respuestas normales.
El estudio fundoscópico directo, el indirecto y los estudios mediante
angiogramas con fluoresceína permitieron detectar en 1os 10 casos afectados
maculopatías de diversos grados, compromiso variable (de moderado a severo)
del epitelio pigmentario retinal, y en seis pacientes diversos grados
de atrofia óptica.
Como casos de observaciones muy especiales, hubo dos pacientes en condición
de franca y grave ataxia cerebelosa (IV-54) y (IV-57) asociados a severas
alteraciones oftalmológicas en relación con el resto de los trastornos
neurológicos.
Debemos destacar el caso del paciente IV-54. Se trata de un varón de 23
ańos de edad (caso índice) con historia de perturbaciones en la marcha
y alteraciones visuales de ocho ańos de evolución. En abril de 1998, se
hallaba imposibilitado para mantenerse en posición de pie, debió permanecer
postrado en cama, tenía grave incoordinación de los cuatro miembros, marcado
temblor intencional, disartria, disfagia, espasticidad generalizada, hiperreflexia,
signos de Hoffmann y Babinski bilateral. No percibía visualmente la luz,
y mostraba una grave palidez de los nervios ópticos, atenuación de los
vasos retinales, maculopatía atrófica, hemorragias en la retina, vasculitis
y atrofia retinal.
La segunda observación (IV-57) (Figs. 2A, B, C y D)
corresponde a un varón de 36 ańos de edad, fue examinado hace 18 ańos.
En aquella oportunidad su agudeza visual fue de 20/200, y el estudio con
angiofluoresceína, así como el electroretinograma permitió diagnosticar
el estado oftalmológico como enfermedad de Stargardt con fundus flavimaculatus
y respuesta electroretinográfica anormal. Actualmente., es uno de nuestros
pacientes con mayor compromiso neurológico y oftalmológico. No existe
percepción visual, tiene compromiso generalizado del epitelio pigmentario
retinal, atrofia óptica, y atenuación de vasos retinales, indistinguible
de una retinitis pigmentosa atípica avanzada, con la maculopatía distrófica
(verificada por estudio angiográfico con fluoresceina) y, además, se encuentra
en oftalmoplejía total. Todo ello evidencia el carácter progresivo del
proceso en alguno de los casos.
En resumen, los pacientes con compromiso oftalmológico presentaron, en
general, maculopatías atróficas, degeneración del epitelio pigmentario
y grados variables de atrofia óptica.
Los tests de matices (Hue test) fallaron para detectar casos precoces.
En casos moderados mostraron sólo algunos errores sin indicar eje de error
y en casos avanzados no se concedió valor a la prueba por el defecto visual
considerable.
Probablemente la perimetría computarizada sea el indicador más precoz
en los pacientes que se inician con compromiso del nervio óptico, ya que
en ellos no pudo detectarse maculopatía con fluoresceína.Se analizó el
ADN de 30 individuos (10 afectados y 20 no afectados) para comprobar la
expansión repetida de los tripletes CAG en el brazo corto del cromosoma
3p (Launce Gouw), obteniéndose resultados definitivos después de cuatro
ańos (1995-1998). Así, el ADN fue identificado, para aspectos de la investigación,
como correspondiente a la familia 2905 y los resultados figuran en la
Tabla 1. En relación con los diez pacientes afectados,
se observó expansión de los tripletes CAG, en todos ellos (Tabla
2).
Como consecuencia de estos resultados se pudieron obtener las siguientes
observaciones:
- En todas las generaciones, los individuos afectados presentaron expansión
de los tripletes CAG en cifras superiores a los intervalos de poblaciones
estudiadas en donde se han encontrado familias con AEC7, con un número
mayor de 37 repeticiones CAG, mientras que los intervalos normales en
poblaciones estudiadas de diferentes nacionalidades están entre 4-17.
El número de repeticiones más frecuente es 10. Un número superior a 37
repeticiones CAG se vincula con la enfermedad.
| TABLA
2 |
| CARACTERISTICAS
GENETICAS de DIEZ PACIENTES AFECTADOS |
| ID |
EDAD |
SEXO |
GENERACION |
ADN
ID |
REPETICION
CAG |
AA
PG
PG
IC
VS
CE
GM-X
FO
EO
JA |
61
35
33
24
57
15
23
36
22
10 |
F
F
M
F
F
F
M
M
F
M |
II-4
III-16
III-17
III-19
III-26
IV-41
IV-54
IV-57
IV-60
IV-48
|
22808
22809
22810
22873
22865
22818
22814
22866
22868
22875
|
44
47
47
47
42
51
59
53
48
63
|
- En la familia investigada
se observa el fenómeno de anticipación genética (16) de una generación
a la siguiente. El fenotipo se presenta más precozmente y la evolución
de las manifestaciones clínicas es más rápida y de mayor gravedad en las
últimas generaciones. De otro lado se conoce la existencia de una inestabilidad
elevada en la expansión repetida de los tripletes CAG. Sin embargo, los
resultados de la clonación del gen para la AEC7 tiene una tendencia un
tanto más estable, con una propensión a mayor número de tripletes CAG
en las generaciones sucesivas de individuos afectados, tal como se ha
podido comprobar en la presente investigación.
Discusión
El pedigree estudiado permite comprobar que se trata de una familia afectada
por una enfermedad autosómica dominante ver (árbol genealógico).
No fue posible precisar por la información de la historia clínica si fue
el compromiso de la visión o el trastorno del equilibrio, el síntoma que
precedería al desarrollo de las otras manifestaciones neurológicas acompańantes
en los pacientes investigados. L.G.Gouw en su trabajo intitulado "Investigación
clínica, neuropatológica y genética en una familia afectada por ataxia
cerebelosa autosomica dominante con degeneración retiniana", destacaron
como resultado de su estudio que la alteración en la discriminación de
los colores, puestos en evidencia mediante el test de Farnsworth (D15),
sería el predictor clínico del inicio de la enfermedad. En nuestra investigación
oftalmológica, este hallazgo anteriormente mencionado no se encontró,
adicionalmente mediante estudios con angiofluorsceina y perimetría computarizada,
este último procedimiento podría revelar la precocidad del defecto del
nervio óptico en ausencia de maculopatía (C. A.).
Adicionalmente podemos afirmar que la disfunción cognitiva encontrada
en los pacientes investigados se ha comunicado en este tipo de ataxia
en estudios previos. De otro lado este aspecto nos obliga a comentar sobre
los nuevos conceptos de la función del cerebelo y la cognición (17). Cada
vez se tiene más evidencia, en animales experimentales, en pacientes humanos
y en estudios de imagen funcional, de la participación del cerebelo en
la función cognitiva (18, 19). Los estudios de imagen funcional muestran
no sólo la activación del cerebelo en tareas puramente mentales sin componente
motor, sino que también seńalan conceptos funcionales novedosos como el
cambio de la activación basada en el aprendizaje de las tareas (20, 21).
Las manifestaciones clínicas descritas por nosotros se relacionaron con
los resultados de los estudios con neuroimágenes (TAC, RMN), donde se
comprobó la considerable atrofia del cerebelo, el tronco encefálico y
de los hemisferios cerebrales., Así mismo, se reiteraron las características
observadas por otros investigadores al considerar esta condición, en término
patológicos, como atrofia olivopontocerebelosa (22).
La investigación genética dio como resultado la expansión anómala de los
tripletes CAG en el brazo corto del cromosoma 3 (3p), situando a esta
enfermedad dentro del grupo de las ataxias espinocerebelosas tipo 7 (AEC
7). El mayor número de expansiones anómalas CAG se encontraron en las
generaciones últimas, guardando una relación directa con la gravedad y
edad de inicio de la enfermedad. Es el caso de mencionar al paciente probando
(IV-54) en quien el estudio molecular determinó 59 expansiones CAG y en
el caso del paciente (IV-57), 53 repeticiones CAG. De máximo interés resultó
la investigación clínica y genética en un nińo de 10 ańos de edad (IV-48),
que en los últimos 2 ańos, se queja de caídas frecuentes cuando efectúa
carreras durante la actividad física en su colegio, el examen neurológico
casi es negativo en la búsqueda convencional de la ataxia, no se queja
de molestias visuales, aún no se ha detectado maculopatía, y sin embargo,
el número de tripletes CAG es de 63, el mayor número encontrado de todos
los pacientes investigados. Es de esperarse que la progresión de la enfermedad
en este caso, sea mayor y su gravedad por igual, quizás observable en
los siguientes ańos, puesto que los integrantes de esta familia afectada
por AEC7, se encuentran bajo vigilancia clínica domiciliaria permanente
(M. A. C.).
El resultado de esta investigación (AEC7), en el aspecto genético molecular
comprende la expansión anómala de los tripletes CAG, y en la misma forma
en las copias de glutamina en individuos afectados, como suceden en otras
enfermedades hereditarias neurodegenerativas, tales como la enfermedad
de Huntington, otras ataxias espinocerebelosas (AEC1, AEC2, AEC3 o Enfermedad
de Machado Joseph, AEC6, la atrofia dentadorubropalidoluysiana o Enfermedad
de Haw River Smith y la atrofia muscular bulboespinal). La mutación subyacente
en estas neuropatologías es una expansión anómala de copias de glutamina
en individuos afectados (23, 24).
El genotipo de nuestra familia corresponde a la AEC7, con defecto del
locus en el cromosoma 3 (3p), como se dijo anteriormente, y su expresión
fenotípica demostró otra característica común, es decir, el fenómeno de
anticipación generacional.
En conclusión, se ha efectuado un estudio clínico y genético molecular
en una familia peruana afectada por ataxia espinocerebelosa tipo 7, ligada
la mutación al cromosoma 3 (3p); constituyendo este reporte el primero
en el Perú y en Latino América, puesto que la única comunicación conocida
sobre una extensa investigación clínica sobre ataxia espinocerebelosa
autosómica dominante, no vinculada a la AEC7, fue descrita hace casi una
década por G. Orosco Díaz y colaboradores al estudiar varias familias
afectadas en Holguín, Cuba (25), cuyo estudio cromosómico fue realizado
posteriormente por Gispert, S. (26) y que permitió calificar a las familias
afectadas pertenecientes a la ataxia espinocerebelosa tipo 2 (AEC2), identificando
el defecto, en el locus genético 12q23.24.
Agradecimiento
Los autores de la presente investigación, desean manifestar su profundo
agradecimiento a los Doctores: Louis Ptácek y Launce G. Gouw de los Departamentos
de Neurología y Genética Humana de la Universidad de Utha, Salt Lake City,
por los estudios de genética molecular. Igualmente nuestro agradecimiento
a la Dra. Eva Klein, Jefe del Departamento de Genética del Instituto de
Salud del Nińo por su asesoramiento y apoyo. Al Sr. Carlos Delgado, Técnico
del Laboratorio del Hospital 2 de Mayo, por el soporte técnico del trabajo.
Bibliografía
1.Rosenberg, R.N. Autosomal dominant cerebellar phenotypes: The
genotype will settled the issue. Neurology. 1990;40:1329-1331.
2. Harding, AE. The clinical features end classification of the
early onset autosomal dominant cerebellar ataxias. Brain 1982;105:1-28.
3. Harding, AE. Clinical features and classification of the inherited
ataxias Adv Neurol 1983; 61:1-14.
4. Rosenberg, R.N. Autosomal Dominat Cerebellar phenotypes: the
genotype has settled the issue. Neurology. 1995;45:1-5.
5. Orr, HT; Chung, MY;Banfi, S. et al. Expansion of an unstable
trinucleotide CAG repeat in spinocerebellar ataxia type 1: Nat Genet 1993;4:221-226.
6. David, G; Abbas, N; Stevanin, G. et al. Cloning of the SCA7
gene reveals a highly unstable CAG repeat expansion. Nat Genet 1997;17:65-70.
7. Flaning, K; Gardner, K. et al. Autosomal dominat spinocerebellar
ataxia with sensory axonal neuropathy (SCA4): clinical description and
genetic localization to chromosome 16q22.1 Am J Hum Genet 1996;59:392-339.
8. Ranum, LP; Schut, LJ; Lundgren, JK; Orr, HT. and Livingston DM.
Spinocerebellar ataxia type 5 in a family descended from the grandparents
of President Lincoln maps to chromosome 11. Nat Genet 1994;8:280-284.
9. Gouw, LG; Digre, KB; Harris, Cp. et al. Autosomal dominant cerebellar
ataxia with retinal degeneration:clinical, neuropathologic and genetic
analysis of a large kindred. Neurology, 1994; 44:1441-1447.
10. Gouw, LG; Kaplan, CD; Haines, JH. et al. Retinal degeneration
characterizes a spinocerebellar ataxia mapping to chromosome 3p.Nat. Genet.
1995;10:89-93.
11. Benomar, A; Krols, Lstevanin, G. et al. The gene for autosomal
dominant cerebellar ataxia with pigmentary macular distrophy maps to chromosome
3p12-p21.1. Nat Genet. 1995; 10:84-88.
12. David, G; Abbas, N; Stevanin, G. et al. The gene for autosomal
dominat cerebellar ataxia type II is located in a 5-cM region in 3p12-p13:
genetic and physical mapping of the SCA7 locus. Am. J.Hum. Gent. 1996;
59:1328-1336.
13. David, G; Durr, A; Stevanin, G. et al. Cloning of the SCA7
gene reveals a highly unstable CAG repeat expansion. Nat Genet. 1997;
17:65-70.
14. David, G; Durr, A; Stevanin, G. et al. Molecular and clinical
correlations in autosomal dominant cerebellar ataxia with progressive
macular dystrophy (SCA7) Hum. Mol. Genet. 1998;7;165-70.
15. Johannson, J; Forgsgren, L; Sandgren, O. et al. Expanded CAG
repeats in Swedish spinocerebellar ataxia type 7 (SCA7) patients: effect
of CAG repeat lenght on the clinical manifestation. Hum, Mol Genet. 1998;
17:65-70.
16. Gouw, LG; Castańeda, MA; Mckenna, Ck. et al. Analysis of the
dynamic mutation in the SCA7 gene show marked parental effects on CAG
repeat transmission. Hum Mol Genet 1998;7:525-32.
17. N. Arriada Mendicoa, E. Otero Siliceo y T. Corona Vásquez. Conceptos
actuales sobre cerebelo y cognición. Revista Espańola de Neurología.1999;29(5):11.
18. Molinari, M; Leggio, MG. et al. Cerebellum and procedural learning:
evidence from focal cerebellar lesions. Brain. 1997;120;1753-62.
19. Gadian, DG; Isaacs, EB; Cross, JH. et al. Lateralization of
brain function in childhood revealed by magnetic resonance spectroscopy.
Neurology.1996;46:974-977.
20. Akshoomoff, N. and Courchesne, E. A new role for the
cerebellum in cognitive operations. Behav Neurosci. 1992;106:731-738.
21. Silveri, MA; Leggio, MG. and Molinari, M. The cerebellum contributes
to linguistic production: a case of a grammatical speech following a right
cerebellar lesion. Neurology. 1994; 44:2047-2050.
22. Weiner, LP; Konigsmark, BW; Stoll, J Jr. and Magladery, JW. Hereditary
olivopontocerebellar atrophy with retinal degeneration: report of a family
trough six generations. Arch Neurol. 1986; 16:364-76.
23. Trottier, Y; Lutz, Y; Stevanin et al. Polyglutamine expansions
as phatological epitope in Huntington's disease and four dominat cerebellar
ataxias. Nature.1995; 378:403-406.
24. Kawakami, H; Maruyama, H; Nakamura, S. et al. Unique features
of the CAG repeats in Machado-Joseph disease. Nat.Genet. 1995;9:344-345.
25. G, Orozco Díaz, MD; A. Nodase Fleites, MD;R. Cordoves Sagaz MD,
and G. Asburger, MD. Autosomal dominant cerebellar ataxia: Clinical
analysis of 263 patients from a homogeneus population in Holguin, Cuba.
Neurology. 1990; 40:1369-1374.
26. Gispert, S; Twell, R; Orozco, G. et al. Chromosomal assigment
of the second (Cuban) locus for autosomal dominant cerebellar atrophy
(SCA2) in human chromosome 12q23.24. Nature Genet. 1993; 4:295-299.
(*) Departamento
de Neurología, Hospital Nacional "Dos de Mayo", Lima - Perú. Facultad
de Medicina, Universidad Nacional Mayor de San Marcos. (**) Centro Oftamológico
Miraflores, Lima - Perú.
|