|
|
Sarcomas de tejidos blandos en niños.
Dres. Luis León A. (*) Resumen Los niños hasta 14 años también pueden tener cáncer; pero la frecuencia y tipo de neoplasia que los afecta, es diferente respecto a los mayores, pues en la edad pediátrica entre leucemias, linfomas, tumores cerebrales y médula espinal suman 2/3 de los casos vistos (en ambos sexos), mientras que en los adultos éste grupo de tumores (también en mujeres y hombres) representa una minoría. Los Sarcomas de Tejidos Blandos en la niñez ocupan el cuarto puesto, ubicación muy importante si consideramos que fuera de esta edad se ubican en el vigésimo lugar. En el Instituto Especializado de Enfermedades Neoplásicas, los portadores de sarcomas de tejidos blandos acuden con tumores en etapas muy avanzadas de su enfermedad, que se traduce en pobres resultados que se obtienen tanto en lo que respecta a recurrencias como en sobrevida. En este trabajo, desarrollado en el Departamento de Investigación, Docencia y Atención quirúrgica en Senos y Tumores Mixtos (STM) del INEN de 1994 a 1977, además de ofrecer los resultados alcanzados con el estudio de 59 niños enfermos con Sarcomas de Tejidos Blandos, 29 del tipo rabdomiosarcoma (de manejo médico básicamente),y 30 no rabdomiosarcoma (que son tributarios de prioritaria alternativa quirúrgica), hacemos una revisión de sus aspectos epidemiológicos, clínicos, patológicos, de diagnóstico y tratamiento en ésta infrecuente pero apasionante patología dentro del ámbito oncológico pediátrico, siendo a veces difícil realizar un adecuado diagnóstico incluso para un patólogo experto en su manejo. Palabras clave: Sarcomas de Tejidos Blandos, cáncer en los niños, tumores pediátricos. Summary Infants, age 0-14 also develop malignant tumors and it's frequency, and the diseases that affect the among us, in Metropolitan Lima, are different than in adults because the sum of leukemia, lymphoma and brain and spinal cord tumors represent 2/3 of the malignant disease, while in adults it represents a minimal amount. However, soft tissue sarcoma in infants stand in 4th place, very import place if we consider that in adults they stand in 20th place. For parents it is a real nightmare to know that one of their kids has cancer, with the particularity that at Instituto de Enfermedades Neoplasicas the carries of this pathology of soft tissue sarcoma attend with tumors in a very advanced stage of the disease, which brings bad results obtained recurrency or whether survival. Besides offering the results reached with the research of 59 ill infants with soft tissue sarcoma, 29 of the rhabdomyosarcoma, and basically 30 non-rhabdomyosarcoma which are tributary of priority surgical alternative, of this infrequent disease, perhaps the most fascinating in the oncological ambit, also the hardest to obtain an adequate diagnostic even for the most expert pathologist in its care. Key words: Soft tissue sarcoma, cancer in infants, pediatric tumors. Introducción Independientemente a sus convicciones religiosas, intelecto y estatus socioeconómico, el peor sufrimiento en la vida de todo padre y madre ocurre en el momento en que un médico le confirma que su niño o niña ha contraído cáncer. Probablemente al inicio de su reacción lo traicionen sus emociones, quizás se desconcierte o tal vez se puede exaltar, injuriar é incluso pensar que puede venir lo peor. El niño que es comprometido por un cáncer, presentará también una serie de vivencias negativas en relación a su enfermedad y tratamiento, siendo primordial el apoyo que se brinde al entorno familiar, pues existe la posibilidad de llegar a desestabilizar la estructura y dinámica interna no sólo familiar, sino también de sus relaciones extrafamiliares, como el colegio y amigos; siendo indispensable una adecuada comunicación con el grupo de especialistas tratantes del niño, médicos, sicólogos, enfermeras y otros. Los niños hasta 14 años (a veces la edad pediátrica se estima 15), al igual que los adultos aunque en menor número, también padecen de tumores malignos y de estos los más comunes, son primeramente las leucemias alrededor del 30%; los tumores del cerebro y sistema nervioso (comprendido el neuroblastoma) 20%; los linfomas ocupan el tercer lugar con 10%; le siguen las neoplasias del riñón (prácticamente todos representados por el tumor de Wilms) y de la pelvis renal (7%); luego los sarcomas de los tejidos blandos y de los huesos entre 4 y 5% cada uno; en los ojos, (casi exclusividad del retinoblastoma) 3.5%, igual al aparato digestivo, luego el sistema respiratorio, genital, urinario y otros. Esto quiere decir que la distribución histopatológica de las neoplasias malignas en niños, difiere mucho cuando se le compara con los mayores (1), en quienes más del 80% incluye a carcinomas y adenocarcinomas de mamas, del aparato digestivo, órganos respiratorios, genitales, sistema urinario, que en conjunto sólo corresponde a menos del 10% en la niñez. En adultos, sumando leucemias, linfomas y tumores del sistema nervioso, apenas significa el 6.5% del total, mientras que en niños llegan al 60%. La mortalidad infantil tanto a nivel global como en nuestro medio es similar a la frecuencia en cuanto a tipos de STB se refiere. La siguiente tabla 1, confeccionada por el Registro de Cáncer de Lima Metropolitana (1990-1993) (2), muestra la frecuencia de cáncer en niños.
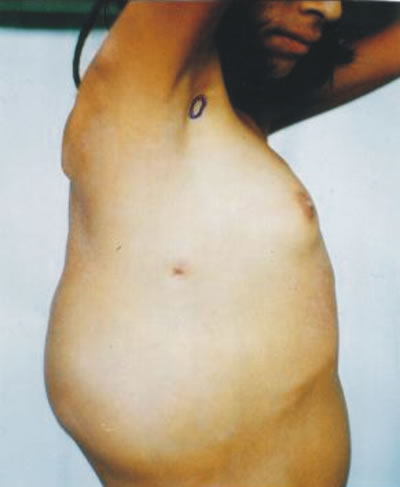
Los tumores malignos de tejidos blandos son llamados sarcomas, pueden aparecer en cualquier tejido blando del cuerpo humano que tenga esta denominación; representan un grupo heterogéneo por su gran diversidad morfológica. Las partes ó tejidos blandos se originan en el mesénquima y son descritos como los tejidos extraóseos, no epiteliales del organismo, con excepción del sistema retículoendotelial, la glía y los tejidos de sostén de algunos órganos parenquimales y vísceras huecas. Por convención (es decir por acuerdo médico universal) también se incluye dentro del grupo a los derivados del sistema nervioso periférico (que nacen en las células de Schwann), que se generan en el ectodermo, porque clínicamente son masas de tejido blando y plantean problemas similares en su morfología, comportamiento clínico, diagnóstico diferencial y tratamiento. Más bien excluye algunos derivados del mesodermo como los huesos, por ser obvio. Los tejidos blandos sirven para conectar, soportar y envolver otras estructuras anatómicas separadas entre sí, estando localizados entre la epidermis y los órganos parenquimales. Están representados por los músculos, tejido fibroso, grasa, vasos sanguíneos y otros tejidos de soporte del organismo como las fascias, tendones y ligamentos. El término sarcoma deriva de la palabra "SARKOS" que en griego significa tumor carnoso, una característica clínica de los sarcomas de tejidos blandos (STB). Fotografía 1.
Clasificación de los STB en la niñez En niños, los STB se clasifican de dos formas: I.- Tipo rabdomiosarcoma (RMS).- Es un tumor maligno del tejido blando de origen muscular estriado, y es el tipo más frecuente en los niños, se estima en la mitad del total de STB pediátricos, no teniendo una presentación homogénea en la clínica, ni en su patología, y tampoco en su respuesta al tratamiento. El rabdomiosarcoma puede localizarse en cualquier parte del cuerpo, pero el 76% lo hace en tres lugares, alrededor del 36% en la cabeza y el cuello, 21% en el aparato genitourinario, 19% en las extremidades. El restante 24% se prorratea entre tronco, tórax, periné, etc. En la niñez, los subtipos histológicos de RMS, según el Intergrupo de Rabdomiosarcoma (IRS-I), que han sido reportados por Maurer y col.(3), son los siguientes, y están expresados en la tabla 2.
El Rabdomiosarcoma (RMS) en niños tiene tres formas de presentación: a) Embrionario, b) Botrioide y c) Alveolar. a) El RMS embrionario, es un tumor maligno a células pequeñas del músculo estriado, que se presenta en niños de menor edad; ocurre habitualmente en el 50-60% de pacientes portadores de RMS, es decir el más frecuente en la niñez, siendo sumamente raro en adultos y en nuestro medio habitualmente alcanzan gran tamaño. b) El RMS botrioide o botrioideo, llamado así porque expresa muy bien la apariencia microscópica del tumor, que tiene un aspecto polipoideo o de racimo; se localiza sobretodo en la vejiga y el sistema biliar y en realidad es una variedad del RMS embrionario. El RMS botrioide marca el 5-6% en las diversas estadísticas. c) El RMS alveolar aparece en los adolescentes y adultos jóvenes, es del orden del 20% en frecuencia, dentro del total de RMS. Habitualmente se localiza profundamente en los tejidos blandos de las extremidades superiores é inferiores y en el tronco, sobre todo en adolescentes y adultos jóvenes, entre 10 y 25 años de edad. Es un tumor altamente maligno, muy agresivo localmente, regionalmente hacia los ganglios linfáticos (un tercio de pacientes), y a distancia básicamente a los pulmones e hígado (2/3 de enfermos). Foto 2.
Conlleva un pronóstico muy serio, presumiéndose que es una enfermedad sistémica de inicio, porque el 80% de los pacientes con enfermedad localmente controlada, sin quimioterapia tienen enfermedad diseminada. El tratamiento generalmente es multidisciplinario, con una combinación que incluye Vincristina, Actinomycina D, Doxorubicina, Ciclofosfamida é Ifosfamida. Inicialmente con éste tratamiento hay una respuesta aparentemente excelente, pero la enfermedad se reactiva a pesar del tratamiento. La sobrevida a 5 años entre el 20 y 35%. Existe otra clasificación más moderna, de acuerdo a las características morfológicas de sus células las redondas, cuyo manejo primario es distinto al de células fusiformes, médico y quirúrgico respectivamente. Tabla 3.
II.- STB de otros tipos no rabdomiosarcoma, NO RMS.- Engloba un grupo de tumores en que cada uno de ellos es relativamente infrecuente y en su conjunto llegan casi al 50% por ciento de todos los STB; comprende una diversidad heterogénea de subtipos histológicos: -Fibrosarcoma Estudios epidemiológicos recientes hechos en Estados Unidos, demuestran diferencias de frecuencia dentro de los STB NO RMS; el subtipo histológico más común en las distintas series en ese país es el sarcoma sinovial, aunque en algunas es el schwannoma maligno, y en otras el fibrosarcoma, pero en la mayoría de estadísticas el más frecuente es el sarcoma sinovial con cifras que bordean el 30-40%, siendo seguido por el fibrosarcoma cerca del 15%, los tumores neurogénicos malignos, alrededor el 10%, lo mismo que los fibrohistiocitomas malignos casi el 10%. Los tumores neurogénicos comprenden a los shwannomas malignos, sarcomas neurogénicos y neurofibrosarcomas. La siguiente tabla 4 muestra la frecuencia de tipo histológico y localización según Dillon y col.(4).
Sin embargo, de acuerdo a Neifeld y col. (5) y Jenkin y Sonley (6), los fibrosarcomas eran los tumores NO RMS más comunes en los niños, y para Brizel y col. (7) son los schwannomas malignos los más frecuentes en su serie, de acuerdo con la clasificación TNM (8) de la Unión Internacional contra el Cáncer. La localización varía, pero alrededor de 65% se ubica en las extremidades, y de ellas el doble más en las inferiores que en las superiores. El espacio retroperitoneal, que en adultos representa una localización importante de los STB, entre 15-20%, en niños es menos común. Los subtipos histológicos NO RMS, varían según su localización anatómica, pero de todos modos la mayor parte están ubicados en las extremidades, y de un modo general en éste lugar se presentan en estadíos más tempranos que en el tórax por ejemplo, donde lo hacen en etapas avanzadas; más aún, los NO RMS de los miembros superiores acuden con enfermedad localizada. Características de los principales El schwannoma maligno es un tumor del nervio periférico, que puede localizarse en cualquier lugar del organismo, pero los sitios más comprometidos son las proximidades de los miembros y de la pelvis. En los niños la mayor parte se originan en la neurofibromatosis o enfermedad de Von Recklinghausen, son multicéntricos y pueden ser superficiales y pequeños, menores de 5 cms; se presentan con mayor frecuencia en la segunda década de la vida. Fotografía 3.
Los schwannomas malignos pueden ser de bajo y de alto grado; de bajo grado tienen pocas mitosis (menores de 5x10 CGA, o campos de gran aumento), o no tienen figuras mitóticas. En los de alto grado, el recuento mitótico es usualmente alto, más de 5 mitosis por 10 CGA. El sarcoma sinovial es la neoformación maligna del tejido sinovial, que se localiza en el tejido conectivo tenosinovial, bursa, y ligamentos, particularmente en las superficies de flexión, es decir en los tejidos sinoviales que se encuentran a lo largo de las fascias, estructuras periarticulares y articulaciones. Frecuentemente aparecen áreas de calcificación en el tumor, lo que produce una imagen radiológica característica, que sirve mucho para su identificación y diferenciación. Si bien se manifiestan en la tercera, cuarta y quinta década de la vida adulta, en los niños ocurre en la segunda década. Se pueden distinguir dos formas: bifásica y monofásica más agresivo y de mayor tamaño. El fibrosarcoma afecta con más frecuencia a los tejidos blandos que a los huesos. Se presenta en cualquier edad y estadío, más comúnmente en varones. En lactantes es el número uno de los STB no RMS. Se localiza de preferencia en las extremidades inferiores. Hay que diferenciar los fibrosarcomas de bajo grado de malignidad, porque muchas veces resulta difícil distinguirlos de la fibromatosis que es su contraparte benigna, menos agresiva pero localmente muy recidivante. Fotografía 4.
Tratamiento para STB Para los niños portadores de STB NO RMS, el tratamiento más adecuado es la cirugía de resección, que es el método primario. El hecho de que sólo menos de las dos terceras partes de pacientes con tumor NO RMS tengan extirpación completa, implica un alto índice de recurrencia local, mientras que hay reparos respecto al rol de quimio y radioterapia postoperatoria. - El valor de la quimioterapia sistémica adyuvante para el tipo NO RMS no está perfectamente definido, especialmente en lesiones de menor tamaño. - La quimioterapia intraarterial puede ser posterior a los procedimientos de salvamento de miembros, en niños con grandes tumores de tipo no RMS. - Cuando se trata de niños pequeños la radioterapia produce diversas complicaciones, como que detiene el crecimiento de huesos y órganos en desarrollo, y también puede inducir la aparición de sarcomas en huesos, tejidos blandos piel y otros órganos. 1°) Agresividad de STB en niños.- De acuerdo a su carácter de agresividad, hay STB infantiles no RMS que tienen diferente conducta. Tabla 5.
Existen STB de comportamiento biológico menos agresivo, como el fibrosarcoma en lactantes y preescolares, el hemangiopericitoma en lactantes y preescolares menores de un año. Algunos STB no RMS presentan un comportamiento biológico similar, es decir son moderadamente agresivos, como el fibrosarcoma en niños y adolescentes mayores, el schwannoma maligno, liposarcoma, sinovioma maligno, el hemangiopericitoma en niños mayores de un año, fibrohistiocitoma maligno y leiomiosarcoma; el manejo de ellos es prácticamente igual. Existen otros tumores cuyo comportamiento biológico es agresivo como el rabdomiosarcoma alveolar, y el sarcoma alveolar de los tejidos blandos en niños, que se caracterizan por su agresividad, requieren una terapia diferente, siendo el estándar la extirpación quirúrgica completa del tumor primario, y, si ésta no fuera posible, se debe indicar radioterapia y quimioterapia considerando que son tumores de un pronóstico muy pobre, el estándar de manejo debe ser la extirpación quirúrgica completa o radical como procedimiento primario, sobre todo para los tumores más agresivos. Si los márgenes resultaran positivos, entonces hay que intentar el retratamiento quirúrgico agresivo, con el fin de evitar el uso de radioterapia en altas dosis en los lactantes, que genera muchos problemas, especialmente retardo en el crecimiento, por lo que sólo debe emplearse cuando la enfermedad sea completamente irresecable. La irradiación en altas dosis produce morbilidad, sobre todo en lactantes y preescolares, por lo que el cirujano debe realizar el máximo esfuerzo mediante cirugía agresiva, para poder hacer el control local de la enfermedad. En los niños mayores la radioterapia se aplica si no cabe nueva cirugía y en caso los bordes de sección sean positivos microscópicamente. Si la enfermedad residual es grosera, desgraciadamente no hay ningún método que brinde una curación definitiva, inclusive utilizando radio y quimioterapia con multiagentes; entonces la alternativa de la amputación puede ser necesaria. En tumores de pronóstico muy pobre, como los del último grupo, con una corta sobrevida, se recomienda quimioterapia neoadyuvante (muchos investigadores consideran de inicio sistémico también al Rabdomiosarcoma embrionario), ó adyuvante. El curso habitual en estos casos, es la progresión de la enfermedad después de un período de remisión. Hay controversias respecto a quimioterapia adyuvante y neoadyuvante. 2°) Pronóstico en niños con RMS.- El pronóstico de la mayoría de los niños con rabdomiosarcoma embrionario ha mejorado mucho en los últimos años, debido a la buena respuesta tanto a radio como quimioterapia, siendo su expectativa de vida superior en la actualidad. 3°) El pronóstico para un niño NO RMS.- El pronóstico de recidiva local y de sobrevida en un niño con STB NO RMS, está supeditado a una serie de factores como son tipo - subtipo, localización, tamaño, lo adecuado de la resección y la presencia de metástasis, el grado histológico, en donde la indiferenciación expresa peor pronóstico que tumores bien diferenciados y por supuesto el comportamiento biológico de agresividad ya mencionado previamente. Material y métodos El presente estudio es descriptivo, del tipo reporte de casos, y tiene como objetivo analizar a la población de pacientes que acuden al Instituto de Enfermedades Neoplásicas (INEN) con diagnóstico de sarcoma de tejidos blandos, con el fin de determinar la frecuencia de su presencia en nuestro medio, de otros aspectos que lo caracterizan y si es posible identificar algún hallazgo que permita plantear un aporte sobre ellos. Con éste fin, se estudiaron 343 casos de Sarcomas de Tejidos Blandos diagnosticados en el INEN en el período comprendido entre el 1ro. De enero de 1994 y el 31 de diciembre de 1997; de los cuales 59 correspondieron a niños con STB de ambos sexos hasta los 14 años. La información se obtuvo directamente de las historias clínicas respectivas. La clasificación y estadiaje de los diversos sarcomas de tejidos blandos, sigue a la elaborada por la Unión International Contra el Cáncer (UICC) en su 6ta.edición del 2002. Hemos escogido el período 1994-1997, de esta patología, que tanto en adultos como en niños en nuestro medio tienen un alto porcentaje de pacientes perdidos de vista, y para los STB en lo que respecta a evaluación de sobrevida y recurrencias, un año de control representa una cobertura de alrededor del 80%, 2 años alrededor del 95% y también por ser más actualizada la recolección relativamente reciente de datos es más fidedigna y susceptible de corregir errores. Resultados del estudio de Hemos observado 59 casos de Sarcomas de Tejidos Blandos en niños, de un total de 343 casos entre niños y adultos, lo que representa en los niños 17,20%, y en los adultos el 82,20 por ciento. Tabla 6.
El tipo histológico más frecuente es el no rabdomiosarcoma con 30 casos (50.85%), mientras que 29 casos (49,15%) correspondieron al tipo rabdomiosarcomas, de acuerdo a la clasificación TNM de la Unión Internacional Contra el Cáncer en 1997. Tabla 7.
Hemos centrado el estudio de los STB en niños, sobre todo al tipo no rabdomiosarcoma, porque como se sabe el tipo rabdomiosarcoma es de manejo médico, aunque puede ser tributario de cirugía cuando algunos aspectos lo considere necesario, como ocurre en casos de gran tamaño del tumor, en donde la amputación es la primera alternativa. El tipo no rabdomiosarcoma es primariamente quirúrgico, pero puede requerir manejo multidisciplinario según la extensión de la enfermedad. De los STB tipo no rabdomiosarcoma (no RMS), el subtipo schwannoma maligno ocupa el primer lugar con el 40%, siguiéndole el neuroblastoma extraadrenal con el 16,66%, luego el fibrosarcoma con el 10%, lo mismo que el neuroepitelioma y el leiomiosarcoma, y 4 casos, 1 de sinovioma maligno, otro tumor neuroectodermal, un liposarcoma y 1 no clasificable, como se aprecia en la tabla 8.
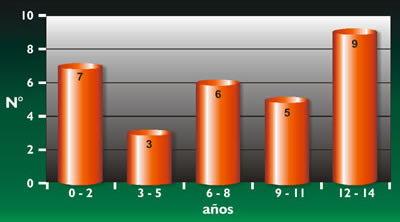
Respecto a la edad en nuestros pacientes de 0 - 14 años, no RMS, vemos (Gráfico 1) que en los últimos 3 años del grupo etáreo en estudio, (12-13-14 años) se presentan la mayor cantidad de pacientes, 9 enfermos. El sexo más frecuente en estos pacientes pediátricos no RMS es el femenino, con el 56,66%. Tabla 9.
La localización de los no RMS: en miembros inferiores 8 casos (26,66%), luego 7 pacientes en el tronco (23,33%), 6 en cabeza y el cuello (20%), 5 en miembros superiores (16,66%) y finalmente 4 en el espacio retroperitoneal (13,33%). Tabla 10. En el retroperitoneo la histología correspondió a dos neuroblastomas extraadrenales, un leiomiosarcoma, y un schwannoma maligno.
El estadío clínico que se vió con mayor frecuencia fue el IV con el 42,85%, luego el III con 23,80% el IIA 14,28%, el IIB 9,52%, y el IA 9,52%, lo que quiere decir que en éste hospital los casos acuden en el 66,66% de las veces en estadíos avanzados de enfermedad. Tabla 11.
En cuanto al grado histológico, en el tipo no RMS, el 64,70% correspondió al G3, 23,52% al G2, 11,73% a G1, significando el 88,22% grados indeferenciados. Tabla 12.
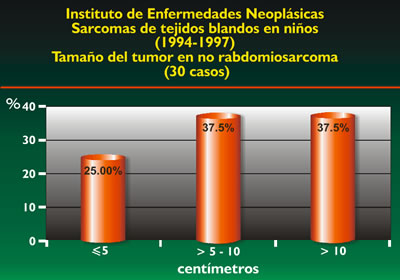
El gráfico 2 nos muestra el tamaño del tumor de acuerdo a los resultados obtenidos, observando que el 75% son mayores de 5 cm.
En cuanto a las recurrencias, la tabla 13 muestra que a los 7 meses han recidivado casi la tercera parte de los niños enfermos.
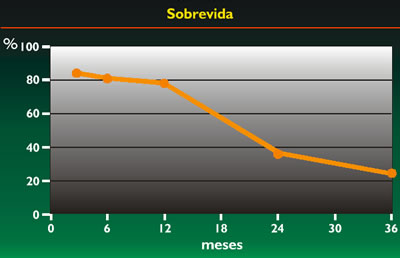
La sobrevida del grupo no RMS se aprecia en el gráfico 3, viendo que es muy pobre, pues más del 80% de niños fallecen a los dos años.
Conclusiones Las conclusiones del estudio acerca de Sarcomas de Tejidos Blandos en niños, según análisis efectuado en el Instituto Especializado de Enfermedades Neoplásicas de 1994-1997 son las siguientes:
(*) Jefe del Departamento de Senos y Tumores Mixtos del INEN. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||