Fisiopatia de la obesidad
Fausto Garmendia Lorena (1)
Regulación de la energía corporal
Con la finalidad de tener un mejor entendimiento de la etiología y fisiopatología de la obesidad, es conveniente efectuar una revisión de los mecanismos de regulación del equilibrio energético corporal total, que se encuentra directa- mente vinculado al mantenimiento del peso; el cual es el resultado de la ecuación:
Ingesta calórica - gasto calórico.
Si la ingesta y el gasto calórico son iguales, se mantiene el equilibrio en cuanto al depósito calórico representado por el peso corporal. En el desarrollo de la obesidad este equilibrio se altera sea por incremento en la ingesta calórica alimenticia o por una disminución del gasto calórico o por la suma de ambos procesos.
La ingesta calórica es fácil de calcular tomando en consideración el valor calórico de los alimentos multiplicados por la cantidad de los mismos. Las proteínas y carbohidratos proporcionan 4 kilocalorías (Kcal) por cada gramo, las grasas 9 Kcal, el alcohol 7 Kcal x gramo; sin embargo los alimentos que se ingiere son mezclas de nutrientes, por lo que es útil recurrir a listas de intercambio, como la preconizada por la American Diabetes Association. Esta institución divide a los alimentos en 7 grupos (Anexo 1).
Para el cálculo del gasto calórico diario se debe considerar el número de calorías utilizadas por los conceptos de:
1) Tasa metabólica de reposo (TMR).
2) Energía utilizada para metabolizar los alimentos.
3) La energía gastada en la activad física, tanto adicional como no adicional.
Las 2 primeras son casi constantes, en tanto que la tercera varía de acuerdo a la cantidad del trabajo físico.
La TMR representa al gasto en condiciones basales, el metabolismo basal corresponde sólo a una parte de la TMR, aunque para fines clínicos son equiparables. El metabolismo basal es el cálculo de la energía corporal, utilizada después de un ayuno de 12 horas, en la mañana y en reposo físico, medida por el consumo de oxígeno en un tiempo determinado. Se reconoce que la TMR tiene variaciones individuales, relacionadas al género, edad, masa de grasa corporal. Una TMR baja es un factor de riesgo para desarrollar obesidad pero no tiene carácter determinante. En el metabolismo de reposo se considera a la energía necesaria para mantener eficientes las actividades funcionales de todos los órganos corporales. En adultos mayores sedentarios el 60% del gasto calórico corresponde al TMR, 10% al gasto térmico (mantenimiento del calor corporal en condiciones de temperatura ambiental intermedia o neutra) y 30% a la actividad física.
En la termogénesis se debe considerar a la necesidad de mantener la temperatura corporal de acuerdo a las variaciones térmicas del medio ambiente (termogénesis adaptativa), así como a la producida por la ingesta alimenticia (termogénesis obligatoria), las proteínas inducen entre 25 a 40% del calor, los carbohidratos 6 a 8% y las grasas apenas 2 a 3%. En conjunto la termogénesis contribuye con alrededor del 10% al gasto calórico total.
En el gasto de calorías por la actividad física se debe tener en cuenta a la adicional (programada) y a la no adicional (no programada), esta última se refiere a la actividad de los movimientos usuales; una persona no baja y sube escaleras en la misma forma, ni utiliza los mismos movimientos ni lo hace con la misma energía que otras. La actividad física programada es la efectuada en forma adicional a las habituales como caminatas, marchas, ejercicios físicos y otros que se imponen por la actividad laboral o deportiva- Levine y col.(1) han planteado que la diferencia en el incremento de peso que se observa en las personas está relacionado a un diferente gasto del componente de la actividad termogénica no vinculada al ejercicio adicional. Esto parece tener importancia para el desarrollo de la obesidad para sujetos en las mismas condi- ciones y se presume que esta relacionado con la actividad del sistema nervioso simpático.
Es evidente que existen diferencias individuales en la capacidad de gastar energía, por las tres vías delineadas anteriormente, metabolismo de reposo, termogénesis y la magnitud de la actividad física que se realice, que explican la facilidad o dificultad para aumentar de peso que tienen los individuos en aparente similar ingesta alimenticia.
Los mamíferos tienen dos tipos de grasa. La grasa blanca que acumula energía en forma de triglicéridos para proporcionar energía en los períodos interprandiales. La grasa marrón con capacidad termógena por disociación del ATP en las mitocondrias por oxidación de los nutrientes que requiere una gradiente de protones, proporcionadas por las proteínas no acopladas (uncoupled protein UCP-1). La cantidad de grasa marrón en el humano es muy pequeña y no tiene mayor función; sin embargo se ha identificado a la UCP 2 y UCP 3 que actúan a nivel del hipotálamo y el tejido muscular.
Etiología
En la actualidad se acepta que la obesidad se produce por diversos factores tanto ambientales que impelen a comer en exceso como por condiciones internas que determinan que el individuo se vea urgido a comer; además intervienen altera- ciones muy precisas que producen obesidad que podríamos llamar secundaria (Tabla 1).
Desde Lavoisier y Laplace en 1783, se considera que existe una regulación fisiológica del balance energético entre la ingesta alimenticia y la pérdida calórica. El desequilibrio energético que se produce en la obesidad es la consecuencia sea de un ingreso energético exagerado frente a una pérdida normal o que con un ingreso normal la pérdida de energía es menor o ambas.
Factores ambientales
La mayor cantidad y disponibilidad de alimentos ofre- cidos por la industria, con gran atractivo gustativo y de alta densidad energética, es un factor importante. Los alimentos ricos en grasas y de alta densidad energética con poco contenido de fibra y de agua inciden más en el incremento del peso, en cambio los alimentos ricos en fibra tienen una densidad baja como las frutas, vegetales y cereales complejos. El otro factor importante es la disminución gradual de la actividad física por diversas causas, muchas de ellas derivadas de los avances tecnológicos, tales como facilidades de transporte, juegos electrónicos, computación, televisión, presión para el estudio y trabajo dentro de una sociedad cada vez más competitiva que disminuye el tiempo para la actividad física y recreativa (Tabla 2).
Factores genéticos o endógenos
En condiciones normales, existe en el organismo un sistema, adquirido en forma genética, que regula el apetito de acuerdo a las necesidades metabólicas del mismo, cuyas alteraciones pueden conducir a la obesidad, que está relacionada a la ingestión de alimentos, las modificaciones metabólicas que ellos originan luego de su absorción intestinal y las señales hormonales que se producen tanto en los períodos prandiales como interprandiales. Este sistema tiene tres niveles de regulación, el sistema nervioso central (SNC), las hormonas del aparato digestivo y el tejido graso.
El hipotálamo es la estructura del SNC con mayor importancia en la regulación de las sensaciones de hambre y saciedad. El neuropéptido Y (NPY) y el péptido relacionado a agouti (AgRP) estimulan el apetito, en tanto que la proopime- lanocorticotropina (POMC), a través de la formación de la hormona melano estimulante α(α-MSH), produce sensación de saciedad.
La ingestión de alimentos provoca en el tracto gastro- intestinal tanto disminución de la motilidad por un mecanismo mecánico como la producción de mediadores u hormonas que frenan el apetito. La colecistoquinina (CCK) se produce en células endocrinas del duodeno y yeyuno en respuesta a la ingesta de grasas y proteínas, actúa tanto a nivel del sistema nervioso central por la vía aferente vagal, como también disminuye la motilidad gastrointestinal. El péptido similar a glucagon (GLP-1 y GLP-2), pertenecen al grupo genérico de las incretinas, participan en la regulación de la glicemia, retardan el vaciamiento gástrico, incrementan la secreción de insulina y disminuyen el glucagon y disminuyen el apetito. El péptido YY, el péptido inhibidor de gastrina (GIP), hormona liberadora de corticotropina (CRH), polipéptido pancreático (PP) y otros polipétidos están implicados en la disminución del apetito (2-5) (Tabla 3). En cambio la Ghrelina, polipéptido estimulador de la somatotropina, que se produce en el estómago, es un estimulador temprano del apetito, su concen- tración plasmática se encuentra en relación inversa al IMC (4).
 |
El tejido graso también interviene en la regulación del apetito a través de la leptina y la insulina, las cuales tienen un efecto inhibidor. En la obesidad, sin embargo, tanto la leptina como la insulina se encuentran incrementadas en sangre, denotando un proceso de resistencia (3).
Una vez que los nutrientes son absorbidos, se produce incremento de glucosa, triglicéridos y aminoácidos. La glucosa se distribuye por varias vías, una parte va al cerebro, donde constituye casi su única fuente energética al lado de los cuerpos cetónicos; otra la lleva a los diferentes tejidos dependientes de la acción de la insulina para constituir fuente de energía o para almacenarse como glucógeno en el hígado y los músculos o para depositarse en el tejido graso como triglicéridos. Estos mediadores bioquímicos también tienen un efecto inhibitorio del apetito.
Después de varias horas de la ingesta alimenticia, el estado metabólico varía en dirección opuesta. Los tejidos dependientes de glucosa como fuente de energía (sistema nervioso), la reciben a partir de la glucogenólisis originada en los dos principales tejidos ricos en glucógeno (hígado y músculo); mientras que la mayoría de órganos y tejidos la obtienen de los triglicéridos y ácidos grasos no esterificados (AGNE), que provienen de la lipólisis incrementada a nivel de los depósitos de grasa. En ese período reaparece la sensación de hambre que está mediada por señales hormonales que también actúan a nivel del hipotálamo, núcleo arcuato, paraventricular y el tronco encefálico. En la tabla 4 se muestran las principales sustancia endógenas estimuladoras del apetito.
 |
Se ha descrito una especie de feedback entre leptina y el neuropéptido Y (NPY), cuando la leptina se incrementa durante la ingesta de alimentos disminuye el NPY y viceversa cuando la leptina disminuye en el ayuno se incrementa el NPY.
Se está investigando la existencia de otros factores genéticos en el desarrollo de la obesidad. Se debe aceptar que el fenotipo de la obesidad es complejo, a diferencia de otras condiciones en las que la ausencia o alteración de un solo locus ocasiona la aparición de un defecto genético (6). Estudios en familias, gemelos, hijos adoptados, pares de hermanos, han permitido conocer que tanto como 80% de las variaciones del IMC están ligados a factores genéticos, que existe transmisión hereditaria en relación a diversos aspectos vinculados a la obesidad, tales como las características fenotípicas de la forma corporal, potencialidad para desarrollar obesidad, distribución de la grasa corporal, actividad física, metabolismo basal, respuesta del gasto energético en relación a la sobrealimen- tación, aspectos relacionados al comportamiento alimenticio, preferencias por los alimentos, actividad de la lipasa lipoproteica, producción de leptina, síntesis de triglicéridos inducida por la insulina, proporción de la lipolisis en el estado basal, entre otros (Tabla 5).
 |
En el momento actual se está investigando sobre genes “candidatos” para explicar la susceptibilidad a desarrollar obesidad que exhiben algunas personas (7,8). Existe una lista importante de estos genes, que se los vincula con aspectos específicos de los mecanismos de desarrollo de la obesidad, como por ejemplo las proteínas de desacoplamiento (UCP), que tienen relación con el gasto energético para generar calor (termogénesis) o el de la producción de leptina, hormona producida por los adipocitos, que tiene relación con la cantidad de la masa grasa y la sensación de saciedad (9,10) (Tabla 6).
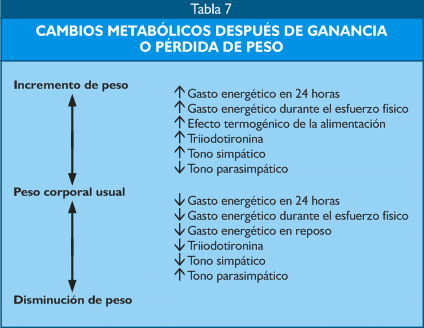
Un fenómeno importante a considerar, particularmente en la dificultad para perder peso, es el que se refiere a las variaciones del gasto calórico cuando se pierde o gana peso, en la primera circunstancia se produce una situación de ahorro energético (Tabla 7).
Obesidad secundaria
Existen condiciones patológicas que producen obesidad (Tabla 8). Desde las psicológicas, en las cuales por estrés psicosocial o estados depresivos, las personas ingieren alimentos en forma compulsiva o como señalan “sin darse cuenta”. Personas en tratamiento psiquiátrico suelen incrementar de peso en forma importante, tanto por la condición patológica como por la terapia con medicamentos que estimulan el apetito.
La región ventromedial del hipotálamo y el tronco encefálico son los sitios del SNC donde llegan las señales aferentes bioquímicas que inducen la sensación de saciedad; por ello lesiones de estos centros están vinculados a incremen-tos rápidos del peso y de la masa grasa.
Enfermedades endocrinas como el síndrome de Cushing, hiperaldosteronismo, hiperinsulinismo provocan incrementos notables de la masa grasa.
Fisiopatología de la patología vinculada
a la obesidad
El estudio y tratamiento de la obesidad ha adquirido importancia por la demostración que es causante de una extensa comorbilidad, es decir que la obesidad favorece la aparición de otras alteraciones, que aceleran el proceso de la ateroesclerosis y la muerte por eventos cardiovasculares (ECV), así como otras condiciones que en conjunto disminuyen la expectativa de vida de las personas. En la tabla 9 se muestran las principales condiciones patológicas que derivan de la obesidad.
 |
En la explicación de estos estados mórbidos, la resistencia a la insulina ocupa un lugar central que da lugar a una extensa cadena de eventos bioquímicos que ocurren en estas alteraciones. El incremento de la masa de tejido graso favorece la liberación de AGNE y del factor de necrosis tumoral α (TNFα), ambos intermediarios bioquímicos inhiben la acción de la insulina (Figura 1). En 1963 Randle y col. elaboraron la teoría del ciclo de la glucosa ácidos grasos, tomando en consideración los resultados de los experimentos in vitro que les permitió demostrar que los ácidos grasos ocasionaban inhibición del transporte y fosforilación de la glucosa a nivel del tejido muscular, esta inhibición del efecto biológico de la insulina estimula la secreción pancreática de insulina (11), a su vez el hiperinsulinismo consiguiente es también otra condición que favorece la resistencia a la insulina por saturación de los receptores de insulina (down regulation).
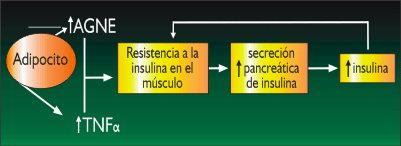 |
| Figura I. Mecanismo de la resistencia a la insulina en la obesidad |
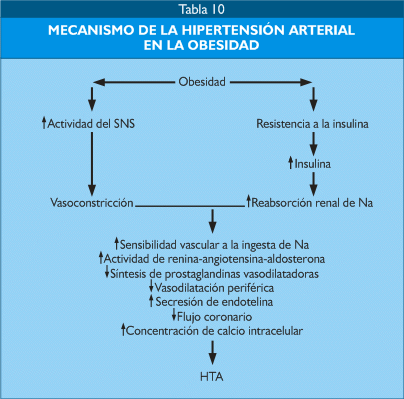
Existe una relación estrecha entre obesidad e hipertensión arterial (HTA), cuyo mecanismo ha sido ampliamente estudiado, como se puede apreciar en la tabla 10; por un lado la obesidad produce resistencia a la insulina e hiperinsulinemia, esta última incrementa la reabsorción renal de sodio y, por otro, el incremento del depósito energético en forma de grasa produce un incremento de la actividad del sistema nervioso simpático. Ambos fenómenos determinan una cadena de alteraciones que al final ocasionan incremento de la presión arterial y, finalmente, hipertensión.
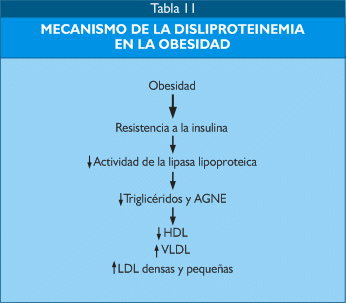
La resistencia a la insulina disminuye la actividad de la lipasa lipoproteica, enzma que favorece la lipogénesis a través de efecto de transferencia de los triglicéridos circulantes a los adipocitos, en consecuencia se produce un incremento de triglicéridos provenientes de las VLDL formadas en el hígado, disminución de HDL e incremento de las LDL, en especial de las partículas pequeñas y densas (Tabla 11).
Hemos señalado que la obesidad provoca resistencia a la insulina e hiperinsulinismo (3), particularmente la de tipo central, abdominal o troncal, por lo tanto, se requiere de una mayor secreción de insulina para mantener la glicemia normal. Ulteriormente se produce daño progresivo de la célula del páncreas que se reflejará primero por un estado de tolerancia disminuida de la glucosa y posteriormente por una diabetes mellitus franca.
La obesidad, hiperinsulismo, dislipoproteinemia, intolerancia a la glucosa y diabetes, hipertensión arterial constituyen factores independientes para desarrollar ateroesclerosis, en consecuencia no debe llamar la atención que los obesos sean más propensos a sufrir ECV.
Las mujeres obesas pueden tener alteraciones menstruales y algunas de ellas sufren de ovarios poliquísticos. El síndrome de ovarios poliquísticos se caracteriza por obesidad, hirsutismo, transtornos menstruales, infertilidad por anovulación y por la formación de múltiples quistes en los ovarios. En estas pacientes se ha demostrado resistencia a la insulina, hiperinsulinismo y tolerancia disminuida a la glucosa o diabetes. En el tejido graso de estas pacientes se aromatizan en mayor proporción los esteroides androgénicos para formar estrógenos, cuyo aporte elevado a la circulación no experi-menta las variaciones cíclicas del ciclo menstrual, lo que es causa de las perturbaciones menstruales y de la anovulación. Se ha demostrado que la administración de metformina a mujeres con ovarios poliquísticos produjo un mayor número de ciclos ovulatorios que las mujeres que tomaron placebo. La metfor- mina es un hipoglicemiante oral que mejora la tolerancia a la glucosa al disminuir la resistencia a la insulina al mismo tiempo que disminuye el hiperinsulinismo. Se puede colegir, que en el síndrome de ovarios poliquísticos la obesidad y la resistencia a la insulina son factores importantes en la anovulación (12). Por otro lado, está muy bien documentado que la elevada producción periférica, no ovárica, de estrógenos incrementa el riesgo de cáncer uterino y mamario en las mujeres obesas.
El exceso de peso y la poca actividad física son las causas más probables de las alteraciones osteoarticulares de los obesos, quienes se quejan de molestias a nivel de la columna vertebral, las rodillas y las articulaciones de la cadera.
La obesidad central disminuye la capacidad respiratoria y al mismo tiempo produce transtornos del ritmo respiratorio durante el sueño, el apnea respiratorio durante el sueño, muy manifiesto en el síndrome de Pickwick, explica la poliglobulia, incremento del volumen sanguíneo y a través de ello la hipertensión arterial. Postulamos que en la exposición a la altura, los obesos tienen una mayor modificación del ritmo respiratorio que las personas de peso normal.
Finalizamos, destacando que la disminución de peso en los obesos demuestra que todas las alteraciones secundarias que se presentan tienen como causa matriz a la obesidad, por que está muy bien documentado que la pérdida de un 10% aún sin llegar a un peso normal, puede ser suficiente para mejorar de las condiciones señaladas anteriormente.
Bibliografía
- Levine JA, Eberhardt NL, Jensen MD. Role of nonexercise activity thermogenesis in resistance of fat gain in humans. Science 1999;283:212-214.
- Stacher G. Saciety effects of cholecystokinin and ceruletide in lean and obese man. Ann N Y Acad Sci 1985;448:431-436.
- Crowell MD, Decker GA, Levy R et al. Gut-Brain neuropeptides in the regulation of ingestive behaviors and obesity. Am J Gastroenterol 2006;101:2848-2856.
- Murphy KG, Bloom SR. Gut hormones in the control of appetite. Experimental Physiology 2004; 89:507-516.
- Woods SC, Seelley RJ, Porte D Jr, Schwarts MW. Signals that regulate food intake and energy homeostasis. Science 1998;280;1378.
- Comuzzie AG, Allison DB. The search for human obesity genes. Science 1998;280:1374.
- Gura T. Uncoupling proteins provide new clue to obesity´s causes. Science 1998;280:1369.
- Marx J. Obesity gene discovery help solve weighty problem. Science 1994;200:1477-1478.
- Zhang Y, Proenca R, Maffei M et al. Positional cloning of mouse obese gene and its human homologue. Nature 1994;372:425-432.
- Sahu A. Leptin signalling in the hypothalamus emphasis on energy homeostasis and leptin resistance. Front Neuroendocrinol 2003;24:225-253.
- Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. Apr 13, 1963;1:785-789.
- Nestler JE, Jakubowizc DJ, Evans WS et al. Effects of Metformin on spontaneous and clomiphene-induced ovulation in the polycistic ovary syndrome. N Engl J Med 1998;338:1876-1880.
1 Profesor Principal, Departamento de Medicina Humana; Investigador Permanente, Instituto de Investigaciones Clínicas; Facultad de Medicina, Universidad Nacional Mayor de San Marcos. |