Actualización sobre la enfermedad de Huntington y experiencia de 30 años en el Instituto Nacional de Ciencias Neurológicas
Luis Torres Ramírez (1), Carlos Cosentino Esquerre (2), Nicanor Mori Quispe (3)
Historia
George Huntington publicó en 1872 un artículo titulado “On Chorea” donde analizaba en una serie de pacientes, el aspecto hereditario de su enfermedad, la presencia y descripción de movimientos anormales, la tendencia a la insania, el deterioro cognitivo, las manifestaciones psiquiátricas y la presentación en la edad adulta (1). Huntington revisó también las notas clínicas de los casos tratados previamente por su padre y abuelo, ambos médicos, a quienes acompañaba en las visitas médicas que realizaban a las familias de East Hampton en Long Island, New York; añadió entonces sus observaciones y escribió el mencionado famoso manuscrito. Por la descripción tan precisa, gráfica, breve y el valioso aporte a la medicina se denominó a esta enfermedad “Corea de Huntington” la cual era conocida simplemente como “corea” (del griego danza) y llamada posteriormente “Baile de San Vito”. En la actualidad el término Enfermedad de Huntington (EH) es el más apropiado para esta entidad.
Definición
La EH se define como un trastorno neurodegenerativo hereditario caracterizado clínicamente por la presencia de movimientos anormales, trastornos psiquiátricos y deterioro cognitivo (2) y que se transmite de manera autosómica dominante, con penetrancia completa, fenómeno de anticipa- ción e inicio en la edad media adulta (3).
En 1983, Gusella et al. (4), encontraron que el gen responsable de la EH se localizaba en el brazo corto del cromosoma 4 luego de estudiar numerosas familias procedentes de Estados Unidos y Venezuela. Diez años después se descubrió el gen IT15 que contenía una mutación caracterizada por una expansión inestable de repeticiones del triplete CAG el que codifica el aminoácido glutamina (5). El gen IT15 codifica a su vez a la proteína llamada huntingtina, cuya función exacta es desconocida hasta le fecha; sin embargo, parece jugar un rol importante en la supervivencia de las neuronas y es esencial para el desarrollo normal antes del nacimiento. Se encuentra distribuida tanto en el sistema nervioso central como en la mayoría de tejidos corporales. En las células, puede ser necesaria para la señalización, el transporte de sustancias, el enlace a proteínas, otras estructuras y proteger a la célula de la apoptosis (6).
Los pacientes con EH poseen la mutación en uno de los alelos del gen conteniendo un número mayor de repeticiones, generalmente más de 40, siendo lo normal inferior a 36. Aquellos con 36 a 40 repeticiones tienen penetrancia incompleta y no necesariamente desarrollarán la enfermedad. Los alelos con más de 28 repeticiones muestran inestabilidad al momento de la replicación celular, la cual se incrementa con el número de tripletes; a mayor inestabilidad esta conduce a la expansión de tripletes, aunque también se ha comunicado la disminución de estos (7). La inestabilidad también es mayor en la espermatogénesis que en la oogénesis, habiéndose reportado los mayores números de repeticiones de trinucléotidos en varones. Este hecho es importante para explicar el fenómeno de anticipación, en el cual la edad de inicio de la EH se torna más temprana en generaciones sucesivas. De la misma forma, los casos nuevos de EH con historia familiar negativa de enfermedad surgen debido a la expansión de un alelo en el límite del rango normal (entre 28 a 35 repeticiones), el cual proviene en la mayor parte de casos del lado paterno. El número de repeticiones de tripletes CAG explica aproximadamente el 60% de la variación en la edad de inicio, con el porcentaje restante representado por genes modificantes y el medio ambiente (8).
Fisiopatología
La EH es causada por una expansión del trinucléotido CAG en el exon 1 del gen que codifica la proteína huntingtina (Htt). La Htt mutante se liga a ciertos factores de transcripción, los cuales por si mismos tienen funciones de poliglutaminas. Actualmente hay dos teorías que podrían explicar la patogénesis de esta enfermedad: desregulación transcripcional y alteración mitocondrial (9).
Sobre la desregulación transcripcional, la Htt se liga a la proteína CBP (cyclic AMP response-element-binding protein (CREB)-binding protein), alterando la expresión de los genes regulados por el factor de trascripción CREB. De esta manera la Htt mutante puede alterar el complemento de proteínas que son sintetizadas en una célula, un cambio que puede conllevar al patrón de neurodegeneración que caracteriza la EH (10).
La otra hipótesis sugiere que la neurodegeneración en la EH resulta de una alteración mitocondrial, ha sido evidenciada a través de estudios bioquímicos, morfológicos y funcionales. La actividad de los complejos transportadores de electrones mitocondriales II, III y IV está reducida en la EH, y la espectroscopía por resonancia magnética del cerebro muestra elevados niveles de lactato, un hallazgo que es consistente con la disfunción mitocondrial, siendo su origen aún un misterio (11).
Una manera de unir ambas hipótesis es gracias al PGC- 1á (proliferator-activated receptor-ã coactivator 1á) peroxisomal, el cual es un coactivador transcripcional que controla varios procesos metabólicos, incluyendo la biogénesis mitocondrial, la fosforilación oxidativa y la termogénesis adaptativa. Además regula la expresión de un amplio número de genes, incluyendo las subunidades codificadas a nivel nuclear de cada complejo de la cadena transportadora de electrones y varios genes que brindan protección en contra de los efectos de las especies reactivas del oxígeno (ROS). Cui et al. (12), reportaron que la Htt mutante reprime la expresión del gen que codifica la PGC-1á mediante la unión a su promotor e interfiriendo con su trascripción dependiente de CREB. Del mismo modo Weydt et al. (13) comunicaron una expresión reducida de los genes regulados por el PGC-1á a nivel del estriado en pacientes con EH, incluyendo las subunidades de la cadena transportadora de electrones. Más aún St.-Pierre et al. (14) demostraron que las células que no expresan PGC-1á tienen un sistema alterado de defensa en contra de las ROS. De esta manera la idea básica en esta enfermedad es que la trascripción del gen que regula la PGC-1á es defectuosa, originando esto una reducción en la expresión de los genes mitocondriales y antioxidantes regulados por la PGC-1α.
Epidemiología
Esta enfermedad se encuentra distribuida en todo el mundo, con una prevalencia de 7 a 10 casos por 100,000 habitantes (15) en población caucásica, comprometiendo ambos sexos por igual y una prevalencia por lo general alta en el norte de Europa (16) aunque se comunican zonas de baja prevalencia (17) , al igual que África (18) y Asia (19).
Cuadro clínico
El inicio de los síntomas es variable y puede desarrollarse desde la primera a la novena década de la vida (20). El promedio de la edad de inicio es entre la cuarta y quinta década, con movimientos involuntarios tipo coreicos en cara y extremidades, que pueden estar ausentes en el reposo; aunque manifestaciones del comportamiento pueden preceder a los movimientos involuntarios en varios años (2,21). Los pacientes se tornan irritables, agresivos, impulsivos y deprimidos (22), posteriormente desarrollan una demencia del tipo subcortical. En esta etapa los pacientes presentan falta de habilidad para mantener la contracción voluntaria (impersistencia motora), alteración de los movimientos sacádicos rápidos en los globosoculares; mas adelante los síntomas motores se generalizan y se añade disartria, disfagia, ataxia, distonía y mioclonías hasta llegar a la postración.
Durante la evolución de la enfermedad se acentúan los cambios de personalidad, apatía, impulsividad, agresión, agitación, depresión, manía, delusiones y alucinaciones. Los cambios cognitivos se manifiestan por pérdida de concentración, memoria, lógica y juicio.
En solo un 10% de casos, si la transmisión de la enfermedad es por línea paterna la enfermedad comienza antes de los 20 años de edad (23) y en ellos puede presentarse un cuadro rígido-aquinético, con ataxia, convulsiones y demencia. El comienzo tardío después de los 50 años ocurre en aproximadamente el 20% de los casos (24).
Diagnóstico
El diagnóstico de la EH se basa en una buena historia clínica que obtenga datos sobre antecedente familiar de enfermedad, síntomas de inicio ya sean motores, psiquiátricos o cognitivos. Luego del diagnóstico clínico, este debe ser confirmado por pruebas genéticas a fin de determinar el número de repeticiones del trinucléotido CAG del gen que codifica la Htt en cada uno de los alelos (25). Debe señalarse que dicha prueba se realiza desde hace varios años en el Instituto Nacional de Ciencias Neurológicas.
Tratamiento
El tratamiento de la EH se basa solo en el control sintomático de los movimientos coreicos y los síntomas conductuales, pues hasta la actualidad no Diagnóstico Tratamiento existe tratamiento para prevenir el inicio de los síntomas o retardar la progresión de la enfermedad. Se están ensayando drogas que muestran un potencial efecto neuroprotector en animales de experimentación (26).
EH en América Latina
Sin duda que el foco más grande de EH en América Latina se encuentra en Zulia al borde el lago de Maracaibo en Venezuela (27). La concentración de pacientes con EH en Zulia representa el más grande foco de casos derivados de un único ancestro común. La alta prevalencia en esta zona fue reportada por primera vez por Negrete (28,29), llamando la atención de la comunidad mundial (30). Las familias afectadas vivían en pequeños centros poblados alrededor del lago y la combinación de aislamiento social y geográfico permitió la expansión de este desorden genético que puede ocurrir cuando el gen es introducido en una población con una tasa elevada de crecimiento. Los otros focos más importantes de EH en América Latina son Cañete-Lima (31) y Cotahuasi-Arequipa (32), ambos en nuestro país.
En el resto de América Latina existen publicaciones de casos aislados de EH (Tabla 1). Los primeros casos fueron comunicados en 1890 y 1891 en Cuba y Brasil por Aróstegui y Cuoto respectivamente, luego en 1894 por Costa en Argentina (27). Gatto et al; publicaron una serie clínica de 11 pacientes argentinos con diagnóstico de EH teniendo como principal síntoma de inicio los movimientos de tipo coreico (33). Cruz- Coke (34), en Chile reportó 10 casos de EH que agregados a los 22 casos previos que comunicaba la literatura chilena hacían un total de 32 casos correspondientes a 12 familias, de las cuales 2 familias con 10 casos eran extranjeras. En Brasil, Barbosa et al; informaron en 1983 las características neuropsiquiátricas, hereditarias y terapéuticas de 16 pacientes con EH (35). Lima e Silva et al. (36), realizaron un estudio molecular en 44 pacientes con EH y en controles con el fin de determinar el tamaño de los alelos expandidos y normales del triplete CAG del gen de la huntingtina. En México, Alonso y Nieto (37) informaban sobre la experiencia en 28 pacientes, correspondientes a 26 familias diferentes, presentándose en todos ellos movimientos coreicos, ya sea al inicio o en algún momento de la enfermedad. Naranjo- Álvarez et al. (38), en Cuba, Reportaron 10 casos de EH de un total de 36 pacientes que provenían de 15 familias, los enfermos tenían un promedio de 48 años con características clínicas similares a las comunicadas en la literatura. Rosselli et al. (39) informaron por primera vez EH en Colombia en 1987, en un estudio que comprendió cuatro familias con 9 adultos y 45 miembros en riesgo de padecer esta enfermedad y a los cuales les realizaron estudios neurológicos y neuropsicológicos. Luego Alonso et al., en 1995 (40) y 1996 (41), escribían sobre su experiencia con pacientes de Juan de Acosta y otras regiones colombianas a los cuales realizaron estudios clínico- moleculares. Más recientemente, Arango-Lasprilla et al. (42), en el 2003 hicieron estudios neuropsicológicos a familias de Antioquia, las cuales habían sido reportadas previamente por Lopera et al. (43).
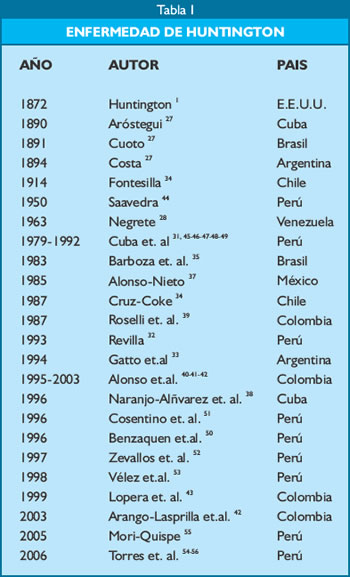 |
EH en el Perú
En nuestro país los primeros reportes datan de 1950 cuando Saavedra (44) comunica el primer caso de EH en un paciente de 39 años, procedente de Cotahuasi - La Unión - Arequipa, con una historia bien documentada de 9 parientes afectados con la misma enfermedad.
En 1979, J. M. Cuba (45), en el Departamento de Neurogía y Gerontología del Hospital “Santo Toribio de Mogrovejo” observó a los primeros pacientes provenientes de la provincia de Cañete situada a 150 Km. al sur de Lima. En colaboración de los doctores Altamirano, Sagástegui, Castro, Benzaquen y Castañeda inician la investigación epidemiológica y la elaboración de los primeros árboles genealógicos de los pacientes con EH. En 1983, Cuba et al. (46), observaron que la edad de comienzo de la EH oscilaba entre los 35 y 45 años, todos descendientes del mestizaje español y establecieron una alta prevalencia, 31 x 100,000, para la provincia de Cañete. En 1986 (31) se realizaron los primeros estudios sobre genealogía de la EH identificando a Cañete como el principal foco de ésta enfermedad en Perú. En 1987, en uno de nuestros múltiples viajes a esa región, tuvimos la compañía de Ira Shoulson, Ann Young, John Penny, Nancy Wexler y otros investigadores de Massachussets-EEUU, quienes realizaron una nueva y completa evaluación clínica, psicológica, obteniendo además muestras de sangre para investigación genética. El grupo peruano del INCN estuvo integrado por Cuba, Torres y Mazzetti así como por médicos residentes y estudiantes del último año de medicina de la Universidad Nacional Mayor de San Marcos.
En 1989 Cuba y Torres (47), comunicaron por primera vez en una revista internacional la descripción de ocho familias con EH en Cañete. Posteriormente en 1990 describen las características clínicas de una gran familia con 30 miembros afectados; esta familia correspondía a una de las l4 estudiadas hasta ese entonces y establecieron que la enfermedad habría aparecido en esa familia entre 120 y 150 años atrás a partir del valle de Cañete diseminándose luego a todo el Perú (48). En 1992 Cuba y Torres (49), publican un caso de ataxia progresiva en una familia con EH.
En 1996, durante la reunión anual de la Academia Americana de Neurología se presentó en un póster otra extensa familia peruana con EH en Latino Americana (50) y ese mismo año en el Congreso de la Sociedad Europea de Neurología se presentó otro póster sobre un estudio abierto del uso de gabapentina en el control del movimiento coreico en la EH (51). En 1997 realizamos un estudio de las impresiones palmares y digitales en pacientes con EH y un grupo control encontrándose diferencias significativas (52). En el V Congreso Internacional de Enfermedad de Parkinson y Desórdenes del Movimiento en 1998 (53) mostramos una casuística de 44 pacientes, de los cuales 37 eran originarios de 10 Departamentos distintos a Lima, lo que demostraba que la EH estaba ampliamente distribuida por el Perú aunque el foco más importante seguía siendo aún Cañete.
En el año 2003 dos de nosotros (LTR-MMQ) y un grupo de estudiantes del último año de Medicina de la Universidad Mayor de San Marcos, viajamos en varias ocasiones a Cañete, para buscar a los descendientes de los primeros pacientes descritos por J.M. Cuba, luego de un seguimiento minucioso elaboramos nuevos árboles genealógicos y se estableció la nueva prevalencia de EH en más de 40 x 100,000 habitantes para el Valle de Cañete, mientras que la prevalencia mundial es de 7-10 por 100,000 habitantes (54-en prensa).
En el 2005, Mori (55) presenta la tesis de Bachiller en Medicina a la UNMSM: ”Epidemiología y Reconstrucción Genealógica de la Enfermedad de Huntington en el Instituto Especializado de Ciencias Neurológicas 1974-2003” elaborada en base a historias clínicas del Departamento de Enfermedades Neurodegenerativas; donde reafirma que el número mayor de pacientes con EH provenían de Lima en especial de Cañete con un 54.4% y que la edad de inicio fue 39.54 años, sin diferencia entre sexos; del mismo modo los pacientes que heredaron la enfermedad por vía paterna tuvieron un inicio más precoz, el mayor porcentaje de pacientes correspondió al inicio en la adultez y que el síntoma inicial más frecuente fue el movimiento coreico.
En el 2006, y en colaboración con el Laboratorio de Neurogenética de nuestro instituto publicamos el “Estudio Clínico Molecular de la Enfermedad de Huntington en Pacientes del Valle de Cañete- Perú” (56), de gran importancia, por ser la primera publicación de pacientes con EH que tienen un diagnóstico clínico confirmado genéticamente.
Durante estas tres décadas, hemos sido testigos y también participes del avance en el conocimiento de múltiples aspectos de esta enfermedad en base a un constante seguimiento epidemiológico, clínico y posteriormente genético que ha permitido confirmar los casos en las últimas generaciones. A través de un protocolo específico en conjunto con las Unidades de Neurogenética y de Movimientos Involuntarios estamos realizando el diagnóstico genético presintomático para la correspondiente consejería genética que es la única medida eficaz para tratar de controlar la diseminación de esta enfermedad.
Bibliografía
- Huntington G. On Chorea. The Medical and surgical reporter 1872;26:317-321.
- Martin JB. Molecular basis of the neurodegenerative disorders. N Engl J Med 1999;340:1970-1980.
- Walker FO. Huntington's disease. Lancet 2007;369:218-228.
- Gusella JF, Wexler NS, Conneally PM. et al. A polymorphic DNA marker genetically linked to Huntington's disease. Nature 1983;306:234-238.
- A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell 1993;72:971-983.
- Bates G. Huntingtin aggregation and toxicity in Huntington's disease. Lancet 2003; 361:1642-1644.
- Djousse L, Knowlton B, Hayden MR. et al. Evidence for a modifier of onset age in Huntington disease linked to the HD gene in 4p16. Neurogenetics 2004;5:109-114.
- Wexler NS, Lorimer J, Porter J. et al. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington's disease age of onset. Proc Natl Acad Sci U S A 2004;101:3498-3503.
- Greenamyre JT. Huntington's disease-making connections. N Engl J Med 2007;356:518-520.
- Landles C, Bates GP. Huntingtin and the molecular pathogenesis of Huntington's disease. Fourth in molecular medicine review series. EMBO Rep. 2004;5:958-963.
- Kwong JQ, Beal MF, Manfredi G. The role of mitochondria in inherited neurodegenerative diseases. J Neurochem 2006;97:1659-1675.
- Cui L, Jeong H, Borovecki F. et al. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 2006;127:59-69.
- Weydt P, Pineda VV, Torrence AE. et al. Thermoregulatory and metabolic defects in Huntington's disease transgenic mice implicate PGC-1alpha in Huntington's disease neurodegeneration. Cell Metab 2006; 4:349-362.
- St-Pierre J, Drori S, Uldry M. et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006; 127:397-408.
- Walker FO. Huntington's Disease. Seminars in neurology 2007; 27:143-150.
- Bates G, Harper P, Jones L. The Epidemiology of Huntington's Disease. In: Harper PS, Eds. Huntington's Disease. London: Oxford University Press, 2002: 251 - 280.
- Palo J, Somer H, Ikonen E. et al. Low prevalence of Huntington's disease in Finland. Lancet 1987;2:805-806.
- Silber E, Kromberg J, Temlett J. et al. Huntington's disease confirmed by genetic testing in five African families. Mov Disord 1998; 13:726-730.
- Leung CM, Chan YW, Chang CM. et al. Huntington's disease in Chinese: a hypothesis of its origin. J Neurol Neurosurg Psychiatry 1992; 55:681-684.
- Pericak-Vance MA, Elston RC, Conneally PM. et al. Ageof- onset heterogeneity in Huntington disease families. Am J Med Genet 1983; 14:49-59.
- Martin JB. Huntington's disease: new approaches to an old problem. The Robert Wartenberg lecture. Neurology 1984;34:1059-1072.
- Haddad MS. Cummings JL. Huntington's disease. Psychiatr Clin North Am 1997; 20:791-807.
- Rasmussen A, Macias R, Yescas P. et al. Huntington disease in children: genotype-phenotype correlation. Neuropediatrics 2000;31:190-194.
- Myers RH, Sax DS, Schoenfeld M. et al. Late onset of Huntington's disease. J Neurol Neurosurg Psychiatry 1985;48:530-534.
- Harper PS, Gevers S, de Wert G. et al. Genetic testing and Huntington's disease: issues of employment. Lancet Neurol 2004; 3:249-252.
- Bonelli RM, Hofmann P. A systematic review of the treatment studies in Huntington's disease since 1990. Expert Opin Pharmacother. 2007; 8:141-153.
- Harper P. Huntington's Disease. Major Problems in Neurology, 2da ed. Londres: Saunders, 1991.
- Negrette A. Corea de Huntington (Estudio de una sola familia investigada a través de varias generaciones), ed. Maracaibo, Venezuela: Universidad de Zulia, 1963.
- Okun M, Thommi N. Americo Negrette (1924 to 2003): Diagnosing Huntington disease in Venezuela. Neurology 2004;63:340-343.
- Young A, Shoulson I, Penney J. et al. Huntington's disease in Venezuela: neurologic features and functional decline. Neurology 1986;36:244-249.
- Cuba JM. [A focus of Huntington's chorea in Perú]. Rev Neurol (Paris) 1986;142:151-153.
- Revilla FJ. Enfermedad de Huntington: Estudio Epidemiológico en un grupo familiar. Tesis de Bachiller en Medicina. Lima - Perú: UPCH, 1993.
- Gatto E, Micheli F, Fernández M. et al. Análisis de una serie clínica: Corea de Huntington. Evaluación Clínica. Rev Chil Neuro-Psiquiat 1994;32:127-128.
- Cruz-Coke R. Epidemiología Genética de Corea de Huntington en Chile. Rev Med Chile 1987;115:483-485.
- Barbosa ER, Marchiori PE, Scaff M. et al. [Huntington chorea: report of 16 cases]. Arq Neuropsiquiatr 1983; 41:191- 198.
- Lima e Silva TC, Guerra H, Bertuzzo CS. et al. Molecular Diagnosis of Hungtington disease in Brazilian patients. Arq Neuropsiquiatr 2000; 58:11-17.
- Alonso ME, Nieto D. Aspectos Clínicos y Genéticos de la Enfermedad de Huntington. Rev Invest Clin 1985;37:125-130.
- Naranjo-Álvarez R, Sherie-Matamoros CE, Salva-Camaño S. Enfermedad de Huntington en Cuba: Estudio Clínico de 10 pacientes. Rev. Ecuat Neurol 1996;5:47-49.
- Rosselli D, Rosselli M, Penagos B. et al. Huntington's disease in Colombia: a neuropsychological analysis. Int J Neurosci 1987;32:933-942.
- Alonso HS, Daza J, Carbonell C. et al. Correlación clínico molecular y caracterización de la Enfermedad de Huntington en familias de Juan de Acosta y otras regiones colombianas. Acta Neurol Colomb 1995; 4:300.
- Alonso HS, Daza J, Carbonell C. et al. Caracterización de las secuencias polimórficas de Tripletes CAG y CCG del gen de la Enfermedad de Huntington en familias colombianas. Acta Neurol Colomb 1996; 12:70-75.
- Arango-Lasprilla JC, Iglesias-Dorado J, Moreno S. et al. Estudio neuropsicológico de la enfermedad de Huntington en familias de Antioquia, Colombia. Rev Neurol(Esp) 2003;37:7- 13.
- Lopera F, Pineda-Trujillo N, Moreno S. et al. Huntington's disease in families from Antioquia, Colombia. Acta Neurol Colomb 1999;15:87-97.
- Saavedra A. Sobre un caso de Corea de Huntington. Rev Neuropsiquiatría 1950;2:232-239.
- Cuba JM. Corea de Huntington: Epidemiología en el Perú y Terapéutica. II Jornada Científica U.P.C.H. Dic 10-15 Lima- Perú Abst. Nº 70. 1979.
- Cuba JM, Castro C, Benzaquen M. Sobre la Epidemiología de la Corea de Huntington en el Perú. Rev Neuropsiquiatría 1983;46:114-120.
- Cuba JM, Torres L. Huit Arbres Généalogiques de Chorée de Huntington au Peróu. Rev Neurol (Paris) 1989; 145:482-484.
- Cuba JM, Torres L. Estudio de una familia con Corea de Huntington en Cañete. Rev Neuropsiquiatr 1990;53:94-102.
- Cuba JM, Torres L. Un cas d'ataxie progressive dans une famille de chorée de Huntington. Rev Neurol (Paris) 1992;148:374 - 376.
- Benzaquen MP, Pachas E, Torres L. et al. Huntington’s Disease in Perú: Another Large Latin-American Kindred. Neurology 1996; 46:PO3-052.
- Cosentino C, Torres L, Cuba JM. Gabapentin for Huntington's disease. J. Neurol 1996;243:P404.
- Zevallos D, Cosentino C, Torres L. Estudio del Angulo Palmar “atd” en la Enfermedad de Huntington. Rev Per Neurol 1997;3:9-12
- Velez M, Cosentino C, Torres L. Huntington Disease in Perú. Mov Disord 1998;13:221
- Torres L, Mori-Quispe N, Mendoza-Cabanillas M. et al. Alta Prevalencia de Enfermedad de Huntington en Cañete- Perú. Parkinsonis Relat Disord 2008 (en prensa).
- Mori-Quispe N. Epidemiología y Reconstrucción Genealógica de la Enfermedad de Huntington en el Instituto Especializado de Ciencias Neurológicas 1974-2003. Lima - Perú: Universidad Nacional Mayor de San Marcos, 2005.
- Torres L, Mori-Quispe N, Mendoza-Cabanillas M. et al. Estudio Clínico Molecular de la Enfermedad de Huntington en Pacientes del Valle de Cañete - Perú. Diagnóstico 2006; 45:102-108.
1 Jefe del Departamento de Enfermedades Neurodegenerativas del Instituto Nacional de Ciencias Neurológicas.
2 Médico Neurólogo, Jefe de la Unidad de Movimientos Involuntarios del Instituto Nacional de Ciencias Neurológicas.
3 Médico Residente de Neurología de la Universidad Nacional Mayor de San Marcos (UNMSM).
|