Sirenomelia
Reporte de un caso
Oscar F. Palacios Carbajal (1), Hugo P. Villate Alvarado (2), Germán Aquino Díaz (3), Cléver Salazar Moreno (4)
Resumen
Es una malformación congénita extremadamente rara; se produce por la fusión de los miembros inferiores secundaria a un trastorno severo en el desarrollo del blastema caudal axial posterior. Se presenta en forma aislada, o asociada (a trastornos renales, cardiovasculares, gastrointestinales, respiratorios, neurológicos y genitales) formando parte del síndrome de regresión caudal. Presentamos el caso de un recién nacido de 38 semanas de edad gestacional hospitalizado en el Servicio Neonatología del Hospital Nacional "Almanzor Aguinaga Asenjo" EsSalud, Chiclayo-Perú portador de Sirenomelia asociada a malformaciones gastrointestinales, genitourinarios y renales, el cual falleció a las 53 horas de vida.
Palabras clave: Sirenomelia, síndrome de regresión caudal.
Abstract
Is an extreme rare pathology. It is produced by inferior limbs fusion, secondary to a severe disorder in the posterior axial caudal blastema development. It presents as an isolated or associated form (renal, cardiovascular, gastrointestinal, respiratory, neurological or genital disorder) constituting part of the Caudal Regression Syndrome. We describe the case of a newborn of 38 weeks, who was attended in the Neonatology Service of Hospital Nacional “Almanzor Aguinaga Asenjo”, EsSalud, Chiclayo-Perú presenting sirenomelia associated to gastrointestinal, renal and genital disorders wich died 53 hours of life.
Key words: Sirenomelia, caudal regression syndrome.
Introducción
La Sirenomelia es una malformación congénita rara, que en la mayoría de casos es incompatible con la vida. Se caracteriza por la presencia de miembros inferiores fusionados, y se asocia con anomalías genitourinarias, renales, gastrointestinales, respiratorias, cardiovasculares y vasculares (1). La incidencia varía desde 1 en 50,000 hasta 1 en 100,000 nacimientos, con una relación masculino-femenino 3:1 (1,2).
Existen varios sinónimos de esta malformación: sirenomelus, monopodia, sirena, simelia, uromelia, feto sireniforme, feto cuspídeo, simpodia, síndrome de Mermaid, síndrome de Vater y anomalía de Duhamel (2,3).
La etiología precisa no es bien conocida y aunque muchas teorías han sido propuestas ninguna se ha considerado concluyente (4).
Stevenson et al. (4) propusieron la teoría de la presencia de una arteria vitelina persistente consecuencia de una alteración en el desarrollo vascular temprano durante el día 22-23 del desarrollo embrionario, así en vez de que la sangre regrese a la placenta a través de las arterias umbilicales pares que se derivan de las arterias ilíacas, la sangre retorna a la placenta, se produce una falta de perfusión, lo que explica alguna de las anormalidades (1) presentes en los fetos sirenomélicos. No se han determinado factores genéticos causales, sin embargo se ha relacionado con la exposición a agentes teratógenos como la vitamina A (a dosis excesiva antes de la 4ta semana del desarrollo), la exposición a cocaína durante la mayor parte del primer trimestre del embarazo y la hiperglicemia en madres diabéticas (1,2,5). Foster citado por Bianchi et al. (6) en 1865 propuso la primera clasificación de sirenomelia según el número de pies: simelia apus casos donde no había pies, simelia unipus cuando había un solo pie y simelia dipus si estaban presentes dos pies. Otra clasificación descrita por Stoker y Heifetz (7) en 1987, se basó en la presencia o ausencia de los huesos de los miembros inferiores.
El objetivo del presente reporte es comunicar el caso de un recién nacido portador de Sirenomelia que ingresó al Servicio de Neonatología del Hospital Nacional “Almanzor Aguinaga Asenjo” EsSalud, Chiclayo-Perú y hacer un aporte al estudio de la sirenomelia como parte del síndrome de regresión caudal.
Presentación del caso clínico
Neonato de 16 horas de vida, sexo no evidenciado que ingresó al Servicio de Neonatología del Hospital Nacional “Almanzor Aguinaga Asenjo” - EsSalud, Chiclayo transferido desde otro centro médico de la ciudad.
Producto de 1era gestación, parto por cesárea por feto en transversa, recién nacido a término (38 semanas de edad gestacional) pequeño para la edad gestacional (1700gr), Apgar 5 y 9 al minuto y cinco minutos respectivamente.
Presentó desde nacimiento dificultad respiratoria que fue incrementándose progresivamente, y múltiples malformaciones congénitas. Madre de 32 años, primigesta, sin controles prenatales, familiares sin malformaciones congénitas.
Al examen físico: frecuencia cardíaca: 148x’, Frecuencia respiratoria: 62x’, Saturación oxígeno: 90%, Peso: 1700 gr.
Recién nacido en mal estado general, en ventilador mecánico, petequias en cuello, abdomen y miembros inferiores, pabellones auriculares de implantación baja. Pulmones roncantes en ambos campos pulmonares. Corazón: ruidos cardiacos rítmicos, regulares. No soplos. Abdomen: globuloso, petequias periumbilicales, RHA escasos.
Miembros inferiores fusionados con presencia de ambos pies con 5 dedos cada uno, pelvis pequeña, pliegue interglúteo ausente y nalgas aplanadas. Agenesia de genitales externos, solo presencia de esbozo dérmico, ano imperforado, ausencia de meato urinario, tono muscular incrementado.
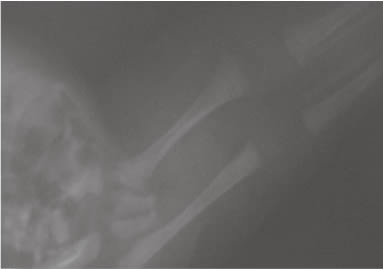 |
| Foto 1. Radiografía de miembros inferiores donde se observa presencia de 2 fémur, 2 tibias y peronés. Clasificación tipo I de Stocker y Heifetz. |
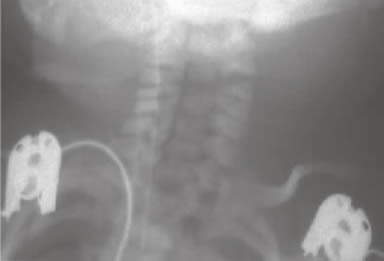 |
| Foto 2. Radiografía cervical donde se observa fondo de saco esofágico compatible con atresia esofágica. |
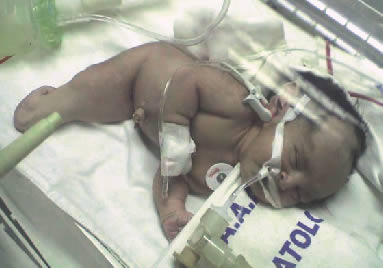 |
| Foto 3. Recien nacido con Sirenomelia Dipus, con miembros inferiores fusionados, dos pies presentes, ausencia de genitales externos y ano imperforado. |
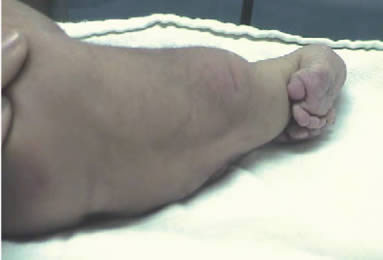 |
| Foto 4. Recien nacido con miembros inferiores fusionados, y dos pies con 5 dedos cada uno. |
Estudios complementarios
• Radiografía toracoabdominal: Fondo de saco esofágico con presencia de aire a nivel intestinal hallazgos compatibles con atresia esofágica con fístula traqueoesofá gico distal.
• Radiografía de miembros inferiores: Se observan 2 fémures, 2 tibias y peronés respectivamente.
• Ecografía abdominal: No se observa vesícula biliar.
En ambas fosas renales no se aprecia imágenes que corresponden a riñones. No hay evidencias de contenido líquido pélvico. Hallazgos en relación con agenesia renal bilateral y ausencia vesical. Ecocardiograma: no evidencia de cardiopatía congénita.
Se plantea por los hallazgos clínicos y por imágenes el diagnóstico de síndrome de regresión caudal: Sirenomelia. Neonato falleció a las 53 horas de vida.
Discusión
La Sirenomelia es una malformación congénita letal extremadamente rara, se puede presentar en forma aislada o formando parte del síndrome de regresión caudal secundaria a una alteración temprana en el desarrollo vascular durante la embriogénesis (4ta semana del desarrollo) con ausencia de las ramas de la parte distal de la aorta abdominal, existe fusión de los miembros inferiores y una amplia gama de anomalías asociadas como producto de un trastorno severo en el desarrollo del blastema caudal axial posterior (3,8).
Esta insólita malformación se define por la fusión de las extremidades inferiores en mayor o menor grado, remedando así la figura de un pez (2). La Sirenomelia es considerada como la forma más severa del síndrome de regresión caudal (9). Se han comunicado alrededor de 300 casos en la literatura mundial (10).
Según la clasificación de Stocker y Heifetz (7), el caso reportado clasificó en el Tipo I: pares de fémur, tíbia y peronés; y según Foster citado por Bianchi et al. (6) como simelia dipus, ambos pies presentes con apariencia de aletas (2 piernas fusionadas y 2 pies).
Las anomalías asociadas a sirenomelia son: presencia de una arteria umbilical única, el sistema gastrointestinal es anormal en todos los casos (1/3 de los casos tienen anomalías del tracto gastrointestinal alto, incluyendo divertículo de Meckel, agenesia de vesícula biliar y atresia duodenal; y en todos los casos hay agenesia del colon terminal y ano imperforado), trastornos del sistema genitourinario (2/3 de los casos tienen agenesia renal bilateral y el resto displasia renal quística, los uréteres y la vejiga están ausentes y cuando se presentan son hipoplásicos), defectos cardiacos (1/4 de los neonatos tienen defectos ventrículo septales), sistema respiratorio presencia de hipoplasia pulmonar casi constante debido al oligohidramnios, y ambigüedad o ausencia de genitales externos (4,7,11,12).
El presente caso además de la fusión de miembros inferiores se caracterizó por la presencia de malformaciones asociadas: atresia esofágica con fístula traqueoesofágica distal, agenesia de vesícula biliar, agenesia renal bilateral y vesical, ano imperforado y ausencia de meato urinario, no se detectaron defectos cardiacos por ecocardiograma.
Los casos reportados en la literatura médica están siempre asociados a muerte perinatal. La sobrevivencia es extremadamente rara y solo es posible en la ausencia de agenesia renal bilateral, cuando los riñones están formados y no son displásicos (4,7).
Su etiología es desconocida, y por ello se han elaborado varias hipótesis: vascular-nutricional, mecánica o defecto mesodérmico que actuarían antes de la 4ta semana. La primera, por disminución de la vascularización caudal; la segunda, por comprensiones; y la tercera por defecto de la migración (2).
El diagnóstico in útero se debe sospechar cuando se encuentra en la ultrasonografia: RCIU, oligohidramnios sin ruptura prematura de membranas, agenesia renal bilateral, alteraciones en las extremidades y arteria umbilical única (6,9). La gestante del caso no se realizó controles prenatales que pudieran haber detectado la malformación in útero. Como procedimientos tendientes aclarar y facilitar el diagnóstico se pueden tomar radiografías en el segundo y tercer trimestre y realizar amnioinfusión para mejorar las dificultades técnicas ecográficas secundarias al oligohidramnios y así observar los miembros inferiores. Se ha informado el uso de la resonancia magnética al feto in útero como otro método diagnóstico complementario (10).
En la actualidad para los neonatos que sufren esta malformación no hay intervenciones fetales que cambien el pronóstico, pues éste depende del compromiso visceral (6).
En los casos donde no exista agenesia renal y el compromiso pulmonar no es severo el tratamiento del recién nacido es quirúrgico con el objeto de corregir las anomalías gastrointestinales, genitourinarias y osteomusculares (6).
El riesgo de repetición es irrelevante, pues no se ha publicado recurrencia familiar. Obviamente la malformación no es transmisible al no sobrevivir el afectado.
Bibliografía
- Solano AF, Saldarriaga W, Isaza C, et al. Foco epidémico de Sirenomelia en Cali, Colombia. Informe de 4 casos en el Hospital Universitario del Valle en 54 días. Colomb Med 2006;37:213-218.
- Rodríguez MA, Carmona de Uzcátegui ML, Chacín PB, et al. Sirenomelia. Rev. Obstet Ginecol Venez 2007;67(3):1-11.
- Pommier J, Martha Eid de, Montero J, Walter H, Hayes D. Sirenomelia. Rev. Inst. Med. 2003;68(122):59-62.
- Stevenson RE, Jones KL, Phelan MC, et al. Vascular Steal: The pathogenic mechanism producing sirenomelia and associated defects of the viscera and soft tissues. Pediatrics 1986;78:451-457.
- Sonek JD, Gabbe SG, Landon MB, et al. Antenatal diagnosis of sacral agenesis syndrome in a pregnancy complicated by diabetes mellitus. Am J Obstet Gynecol 1990;162(3):806-808.
- Bianchi D, Crombleholme T, D Alton M. Sirenomelia. En: Fetology. New York; Mc Graw Hill; 2000;649-655.
- Stocker J, Heifetz S. Sirenomelia, a morphological study of 33 cases and review of the literature. Perspect Pediatric Pathol 1987;10:7-50.
- García BJ, Romero Araus J. Sirenomelia. Ginecol Obstet. Mex. 1986;64:422-429.
- Valenzano M, Paoletti R, Rossi A, et al. Sirenomelia. Pathological features, antenatal ultrasonographic clues, and a review of current embryogenic theories. Human Reprod Update 1999;5(1):82-86.
- Duhamed B, et al. The caudal regression syndrome. J Ultrasound Med 2002;21(8):915 -920.
- Sozubir S, et al. Sirenomelia with esophagoal atresia. Adv. Clin Path 2000;4(4):165-168.
1 Médico Asistente del Servicio de Neonatología del Hospital Nacional "Almanzor Aguinaga Asenjo" (HNAAA) - EsSalud Chiclayo - Perú.
2 Médico Jefe de Servicio de Neonatología del Hospital Nacional "Almanzor Aguinaga Asenjo".
3 Médico Asistente del Servicio de Cirugía Pediátrica del Hospital Nacional "Almanzor Aguinaga Asenjo".
4 Residente del 1er año de Pediatría rotante en Servicio de Neonatología del Hospital Nacional "Almanzor Aguinaga Asenjo". |
