Esclerosis Tuberosa: Experiencia
en pacientes pediátricos en el Hospital Nacional Cayetano Heredia
Dres. Maria del Pilar Medina
(*), Daniel Guillén (*), Iván Espinoza (*), Patricia
Campos (*)
María Elena Liendo (*), María Isabel Quiroga de Michelena
(**)
Resumen
Se realizó un estudio observacional, de tipo serie de casos,
con el objetivo de describir las características clínicas
de los pacientes pediátricos con diagnóstico de Esclerosis
Tuberosa (ET) controlados en el consultorio de Neurología Pediátrica
del HNCH. La edad promedio de los pacientes al momento del diagnóstico
fue de 5.3 años. El motivo principal de consulta fue convulsiones
en un 87,5%. La edad promedio al inicio de los síntomas fue de
11 meses. La tercera parte de los pacientes tuvo historia familiar compatible
con ET en familiares de primer grado. Las lesiones más frecuentes
fueron las máculas hipocrómicas (81%), los angiofibromas
faciales (56,2%), y la piel de Zapa (31%). Tres de los 16 de los pacientes
presentaron únicamente crisis generalizadas, 2 únicamente
crisis parciales y 5 una combinación de ambas. Cinco pacientes
debutaron con espasmos infantiles, a una edad promedio de 5.8 meses.
Los hallazgos tomográficos fueron los nódulos subependimarios
(91.6%) y los tuberomas corticales (31.2%); tres de ellos únicos.
Cuatro pacientes presentaron ambas lesiones. Se presenta el primer reporte
de Esclerosis Tuberosa en niños de nuestro medio, las características
clínicas encontradas coinciden con las descritas en la literatura,
siendo las convulsiones de difícil manejo el motivo principal
de consulta.
Palabras clave: esclerosis tuberosa, convulsiones, niños
Summary
This is an observational study, designed to describe the clinical characteristics
of the pediatric population with tuberous sclerosis complex who attend
our hospital. The mean age at the time of diagnosis was 5.3 years. The
mean age at the beginning of symptoms was 1 month, and the most frequent
symptom of consultation were seizures. One third of the patients had
first degree relatives with diagnosis of tuberous sclerosis complex.
The most common skin lesions were hypomelanotic macules, facial angiofibromas,
and the shagreen patch. Three patients had generalized seizures, two
had partial seizures and five had both types. Five patients presented
infantile spasms at the mean age of 5.8 months. The most common tomographic
findings were subependimal nodules (91.6%) and cortical tubers (31.2%),
four patients had both findings at the same time. This is the first report
of tuberous sclerosis complex in pediatric patients in our country, and
the main clinical features coincide with those reported previously in
medical references.
Key words: tuberous sclerosis complex, seizures, children.
Introducción
El Complejo Esclerosis Tuberosa es una enfermedad autosómica
dominante, de gran expresividad clínica, que compromete múltiples órganos,
siendo el cerebro, la piel y los riñones los más afectados,
en orden de frecuencia. En las poblaciones estudiadas la prevalencia
de esta enfermedad varía entre 1:1000 a 1:10000 personas (1) y
su presentación puede ir desde pacientes libres de síntomas
hasta discapacidad severa.
Los niños afectados inician los síntomas en edad variable
y hasta un 85% presentan síntomas neurológicos, siendo
los más frecuentes la epilepsia, el retardo mental y el autismo
(2).
En nuestro país se desconoce la prevalencia de esta enfermedad
en niños, así como las formas de presentación más
comunes en pacientes pediátricos.
Objetivo
Describir las características clínicas de los pacientes
pediátricos con diagnóstico de esclerosis tuberosa controlados
en el consultorio de Neurología Pediátrica del Hospital
Nacional Cayetano Heredia.
Metodología
Se diseñó un estudio observacional tipo serie de casos.
Se procedió a la revisión de historias clínicas
y la confección de fichas de recolección de datos y seguimiento.
Para el diagnóstico se precisa encontrar dos o más criterios
mayores o un criterio mayor además de dos o más criterios
menores (3) (Tabla 1).
Todos los pacientes con sospecha diagnóstica fueron evaluados
por los Servicios de Dermatología, Genética, Neuropsicología
y Oftalmología. Los resultados de estas evaluaciones se consignan
en la ficha de recolección de datos.
Resultados
1. Descripción de la población
Se presentan 16 niños con diagnóstico de Esclerosis Tuberosa
(ET) con seguimiento en la Consulta Externa de Neuropediatría
del HNCH en el periodo 1997-2003.
La edad promedio de los pacientes al momento del diagnóstico
fue de 5.3 años. El rango de edades fue amplio, siendo el paciente
más pequeño una recién nacida de 7 días de
vida que debutó con arritmia cardíaca atribuida a un rabdomioma
cardiaco, quien además presentó lesiones dérmicas
típicas y más tarde convulsiones.
La población estudiada mostró una distribución
similar en cuanto a sexo, 8 varones, 8 mujeres. El motivo principal de
consulta de estos pacientes fue el manejo de las crisis epilépticas
en un 87,5% (14/16). La edad promedio al inicio de los síntomas
fue de 13 meses, siendo un 68.7% pacientes menores de un año.
Dos pacientes consultaron por lesiones dérmicas, ambos tenían
antecedente familiar de esclerosis tuberosa, y uno ellos presentó convulsiones
poco tiempo después de la primera consulta.
Si bien la enfermedad tiene herencia autonómica dominante, sólo
una tercera parte de los pacientes tuvieron historia familiar compatible
con Esclerosis Tuberosa en familiares de primer grado.
| TABLA
1 |
 |
2. Cuadro clínico
a. Lesiones dérmicas
Todos los pacientes presentaron lesiones dérmicas al momento
del diagnóstico. Las lesiones más comunes fueron las máculas hipocrómicas, (Fig. 1) presentes en el 86% de
los pacientes y encontradas en la totalidad de pacientes con síntomas
antes del primer año.
En 9 pacientes (56.6%), se observaron los angiofibro-mas faciales (Fig.
2), todos estos niños fueron mayores de 5 años de edad
al momento de la primera consulta. La piel de Zapa (shagreen patch),
una lesión tipo placa de piel engrosada que tiende a localizarse
en la zona dorsolumbar, fue vista en 5 de los 15 pacientes. Uno de los
pacientes presentó una lesión compatible con un fibroma
gingival.
b. Síndrome convulsivo
El 93% de los pacientes (un total de 15) presentaron convulsiones. Ocho
de estos 15 niños presentaron únicamente crisis generalizadas,
2 únicamente crisis parciales y 5 una combinación de ambas.
Con respecto a las crisis parciales, la mitad fueron catalogadas como
complejas. Cuatro pacientes (25%) debutaron con espasmos infantiles,
entre los 3 y 7 meses de edad. En dos pacientes se observó el
inicio de las crisis tiempo después del diagnóstico de
ET. Una paciente tuvo el diagnóstico de síndrome de Lennox-Gastaut
a los 3 años de edad, presentando crisis convulsivas de difícil
manejo.
| FIGURA
1 |
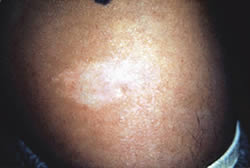 |
| Mancha hipomelanótica
en el tronco de un paciente de 9 años. |
|
| FIGURA
2 |
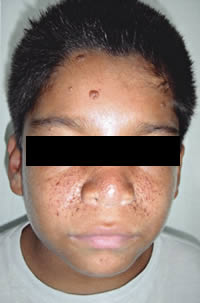 |
| Angiofibromas faciales en un paciente varón
de 10 años. |
|
En un solo paciente el examen neurológico fue normal; en éste
paciente el diagnóstico de ET se hizo en base al antecedente familiar,
las lesiones dérmicas y los nódulos subependimarios detectados
por la tomografía cerebral.
c. Retardo del desarrollo y Retardo Mental
La evaluación neuropsicológica demostró que 15
/ 16 niños tuvieron algún grado de retardo mental o del
desarrollo. En 10 niños se encontró retardo mental, siendo
de grado leve en 8 de éstos, los dos restantes fueron de grado
severo. En los niños con retardo del desarrollo generalmente éste
fue de tipo mixto (motor y del lenguaje) y de grado moderado. Sólo
una niña se encontró con nivel fronterizo con trastorno
de atención e hiperactividad y otro niño con nivel mental
normal (Tabla 2).
3. Neuroimágenes
a. Tomografía cerebral
Todos los pacientes tuvieron tomografía cerebral, siendo los
nódulos subependimarios el hallazgo más frecuente (91.6%).
Seis pacientes tuvieron la asociación de tuberomas corticales
y nódulos subependimarios; (En un solo caso se encontró una
lesión compatible con tuberoma aislado, esta niña tenía
fuertes antecedentes de ET, y además presentaba las máculas
hipocrómicas). En la Fig 3. se muestra los nódulos subependimarios
como lesiones hiperdensas, que pueden ser de tamaño variable.
b. Resonancia magnética
Dos de nuestros pacientes tuvieron RM cerebral. Una de ellas presentó múltiples áreas
frontales y parietales de hiperseñal cortico-subcortical, compatibles
con tuberomas corticales. La RM del otro paciente mostró además
de las zonas de hiperseñal cortico-subcortical, algunas zonas
nodulares periventriculares y una gran zona de hiperseñal en T2
y flair frontal derecha (Fig 4).
Discusión
El Complejo Esclerosis Tuberosa es una enfermedad que resulta de las
mutaciones en los genes TSC1 (cromosoma 9q34) y TSC2 (cromosoma 16p13.3).
La mutación en el gen TSC1 inactiva la proteína hamartina,
mientras que la afección del gen TSC2 alterara la función
de la proteína tuberina. Estas proteínas están ampliamente
distribuidas en el cerebro, riñón, corazón entre
otros. El complejo hamartina - tuberina podría participar en numerosas
vías de señalización celular, incluyendo las que
regulan el crecimiento celular (en respuesta a algunos factores como
insulina), el tráfico intracelular, y la proliferación,
adhesión y migración celulares (4). Asimismo existe evidencia
que indica que ambos genes TSC son supresores tumorales.
* En una niña se encontró un tuberoma certical aislado,
ella tenía antecedentes familiares de ET y además presentaba
máculas Hipocrómicas.
| FIGURA
3 |
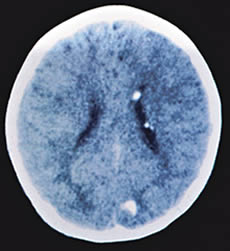 |
| TAC cerebral que muestra nódulos
subependimiarios y tuberoma occipital. |
|
| TABLA 2 |
| CARACTERíSTICAS DE LOS NIÑOS
CON ESCLEROSIS TUBEROSA EN EL HOSPITAL NACIONAL CAYETANO HEREDIA,
2004 |
|
Ficha
|
Edad de consulta
|
Sexo |
Historia Familiar |
Lesiones dérmicas
y mucosas |
Desarrollo y Evolución |
Lesiones SNC |
Edad de inicio de crisis |
Tipo de crisis epiléptica |
| 1 |
3 años |
F |
No |
Piel de Zapa, ibroma ingival |
Retardo mental leve |
Nódulos subependimarios |
16 meses |
Crisis generalizadas, ausencias, crisis
parciales |
| 2 |
3 años |
F |
Si |
Máculas hipocrómicas |
Retardo mental leve |
Tuberoma cortical |
1 año |
Crisis generalizadas |
| 3 |
3 meses |
F |
Si |
Máculas hipocrómicas.
lesiones en confetti |
Retardo motor leve |
Nódulos subependimarios |
3 meses |
Espasmos infantiles |
| 4 |
9 años |
F |
Si |
Máculas hipocrómicas.
angiofibromas, piel de Zapa |
Retardo mental leve |
Nódulos subependimarios |
8 meses |
Crisis paciales y generalizadas |
| 5 |
5 años |
M |
Si |
Máculas hipocrómicas.
angiofibromas faciales |
Retardo mental leve, hiperactividad |
Tuberoma cortical, nódulos subependimarios |
8 meses |
Crisis generalizadas, crisis parciales
complejas |
| 6 |
7 días |
F |
no |
Manchas hipocrómicas |
Retardo motor y del lenguaje,
grado leve |
Tuberomas corticales, nódulos subependimarios |
1 año |
Crisis generalizadas, crisis parciales
complejas |
| 7 |
18 meses |
F |
no |
Manchas hipocrómicas,
angiofibromas faciales |
Fronteriza
Trastorno de atención |
Tuberomas corticales, nódulos subependimarios |
4 meses |
Crisis parciales |
| 8 |
2 años |
F |
si |
Manchas hipocrómicas,
piel de Zapa |
Retardo motor moderado |
Tuberomas corticales, nódulos subependimarios |
13 meses |
Crisis generalizadas, crisis parciales
complejas |
| 9 |
7 años |
M |
no |
Manchas hipocrómicas,
angiofibromas faciales |
Retardo mental leve |
Nódulos subependimarios |
9 meses |
Crisis generalizadas |
| 10 |
5 años |
M |
no |
Manchas hipocrómicas,
angiofibromas faciales |
Retardo mental leve |
Nódulos subependimarios |
7 meses |
Espasmos infantiles, crisis generalizadas |
| 11 |
11 años |
M |
no |
Manchas hipocrómicas |
Retardo mental severo |
Nódulos subependimarios |
2 meses |
Crisis parciales |
| 12 |
8 años |
M |
no |
Angiofibromas faciales |
Retardo mental severo, autismo |
Nódulos subependimarios |
3 meses |
Espasmos infantiles |
| 13 |
12 años |
F |
no |
Angiofibromas faciales, piel
de Zapa |
Retardo mental leve |
Tuberomas corticales, nódulos subependimarios |
4 años |
Crisis generalizadas |
| 14 |
7 años |
M |
no |
Manchas hipocrómicas |
Retardo mental leve |
Nódulos subependimarios |
11 meses |
Crisis generalizadas |
| 15 |
12 años |
M |
Si |
Manchas hipocrómicas,
angiofibromas faciales |
No |
Nódulos subependimarios |
NC |
No |
| 16 |
5 meses |
M |
no |
Manchas hipocrómicas |
Retardo motor y del lenguaje,
grado moderado |
Tuberomas corticales, nódulos subependimarios |
5 meses |
Espasmos infantiles |
|
| FIGURA
4 |
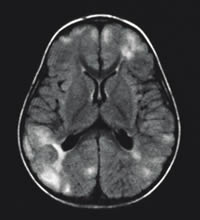 |
| RMN cerebral que muestra múltiples
zonas de hiperseñal en flair de localización
cortico subcortical correspondientes a tuberomas |
|
Un tercio de los pacientes de nuestra serie de casos presentó algún
familiar con esclerosis tuberosa, lo que coincide con la literatura revisada,
que refiere que dos tercios de los casos de ET se atribuyen a mutaciones
espontáneas (5). Asimismo, existe una distribución similar
de mutaciones de TSC1 y TSC2 en los casos familiares, mientras que en
los casos esporádicos la mutación del gen TSC2 es mucho
más frecuente dando como resultado casos más severos, con
aparición más temprana de epilepsia y mayor número
de lesiones cerebrales (6). Se ha descrito además, un riesgo de
recurrencia del 2% en los hijos restantes de padres que no tienen la
enfermedad pero que tienen un hijo con ET por mutación espontánea
(2).
Recientemente se ha asociado la severidad del cuadro clínico
de ET a la capacidad de expresión de los genes TSC1 y TSC2, siendo
este último el implicado en los casos más severos (2).
En la Conferencia Consenso del Complejo de Escle-rosis Tuberosa (CET)
realizada en 1998 (3) se definió 11 criterios mayores y 9 menores,
realizándose el diagnóstico de CET con dos criterios mayores
o con uno mayor y uno menor. También se consideró el diagnóstico
probable con un criterio mayor y el posible con dos o más criterios
menores. Sin embargo, debe resaltarse que algunos de los criterios descritos
no aparecen sino hasta la adolescencia o incluso la edad adulta. Algunos
autores proponen estratificar los criterios mencionados según
edad, haciendo así más sencilla la evaluación a
edades tempranas (7).
Las lesiones cerebrales en ET representan zonas de diferenciación
anormal de las células nerviosas (8), y se clasifican actualmente
en dos tipos, hamartomas y hamartias, siendo necesaria la presencia de
dos hamartomas para hacer el diagnóstico de ET. Hacemos énfasis en la evaluación integral del paciente
neurológico, puesto que el signo más temprano y frecuente
encontrado en nuestro grupo de pacientes fueron las manchas hipocrómicas
en "hoja de fresno", las cuales podrían pasar desapercibidas
sin un examen minucioso de la piel. Otro aspecto de importancia es la
correlación de ciertas lesiones dérmicas y la edad del
paciente, por ejemplo las máculas hipocrómicas pueden encontrarse
a cualquier edad, mientras que los angiofibromas faciales siempre son
lesiones más tardías. En nuestra población todos
los niños mayores de cinco años con ET tenían esta
característica clínica.
En contraste con las lesiones dérmicas, que son claves diagnósticas
sin repercusión clínica, llama la atención que el
cuadro convulsivo no forma parte de los criterios diagnósticos,
sin embargo es la causa más común de consulta en los pacientes,
a menudo por crisis de difícil control (9). Los espasmos infantiles
y las convulsiones parciales con generalización secundaria se
describen como el tipo más frecuente (10), coincidiendo con lo
visto en el grupo de nuestros pacientes.
La ET es una de las causas más frecuentes de Síndrome
de West en la población infantil, aunque se desconoce la razón
de esta asociación. Se han descrito características clínicas
y electroencefalográficas particulares en pacientes con esclerosis
tuberosa, que las diferencian del West clásico (11). Existen además
trabajos que encuentran una asociación entre la presencia de espasmos
infantiles y el número de tuberomas corticales y su localización
(12), generalmente se trata de pacientes que responden mejor al tratamiento
convencional con ACTH y anticonvulsivantes como valproato y vigabatrina.
Con frecuencia se piensa que los niños afectados con ET además
tendrán retardo mental, sin embargo esta asociación sólo
ha sido descrita hasta en el 50% de casos (2). En nuestra serie el porcentaje
fue ligeramente mayor (62.5%), no se ha podido determinar si otros factores
tienen influencia, como por ejemplo: demora en el control de las crisis,
demora en la identificación de la enfermedad, factor nutricional,
factor traumatismo craneal (en los casos con crisis de difícil
control), etc. Por otro lado se conoce que la asociación de retraso
mental y convulsiones es directamente proporcional, y coincide con lo
revisado, todos los pacientes con retardo mental en nuestra serie presentan
convulsiones.
Asimismo se estima que el autismo coexiste en un 17 a 61% de pacientes
con esclerosis tuberosa (13). La incidencia es igual en la población
masculina y femenina con ET, en contraste con la población general,
donde el autismo es cuatro veces más frecuente en los varones.
En el grupo estudiado sólo uno de los pacientes tuvo diagnóstico
de autismo.
Para el diagnóstico de ET la literatura describe con precisión
los hallazgos en RMN (14), incluso se han descrito patrones de lesiones
en RMN específicos para recién nacidos y lactantes (15),
con lo que el diagnóstico temprano en este grupo etáreo
es más preciso. Esta prueba se ha convertido en los últimos
años en la neuroimagen de elección para el diagnóstico
de ET, puesto que permite una localización y definición
mucho más precisa de los tuberomas. Sin embargo, la RMN cerebral
puede no mostrar las lesiones subependimarias patognomó-nicas
de ET. Por tanto la tomografía cerebral sigue siendo en nuestro
medio un instrumento útil y al alcance para el estudio de nuestros
pacientes. Se ha descrito una correlación entre el número de tuberomas
corticales y la existencia de espasmos infantiles, aparición temprana
de convulsiones o retardo mental (16,17) mas en la población estudiada
no se pudieron demostrar tales datos.
El número de pacientes con esclerosis tuberosa y autismo es muy
pequeño para lograr establecer correlación alguna entre
los hallazgos tomográficos y este tipo de presentación
clínica (18), como lo sugieren algunos trabajos recientemente
publicados.
Conclusión
Se describe una serie de casos pediátricos de Esclerosis Tuberosa,
que demuestran la existencia de esta enfermedad en nuestro medio, con
similares características a las descritas en la experiencia internacional.
Aunque las convulsiones no forman parte de los criterios diagnósticos,
son el motivo más frecuente de consulta. Asimismo se insiste en
la importancia de hacer el diagnóstico oportuno por las implicancias
genéticas y de manejo de la comorbilidad.
Bibliografía
- Curatolo, P. Tuberous sclerosis complex. From basic science to clinical
phenotypes. McKeith Press 2003.
- Kandt R. Tuberous sclerosis complex and neurofibromatosis type 1:
the two most common neurocutaneous diseases. Neurol Clin N Am 2002;20:941-64.
- Roach, ES; Gomez, MR; Northrup, H. Tuberous sclerosis complex consensus
conference: revised clinical diagnostic criteria. J Child Neurol
1998;13:624-8.
- Narayanan, V. Tuberous Sclerosis Complex: Genetics to Pathogenesis.
Pediat Neurol 2003;29(5):404-9.
- Crino, PB; Henske, EP. New developments in the neurobiology of
the tuberous sclerosis complex. Neurology. Oct. 1999;53(7):1384-90.
- Janniger, CK; Bielicka-Cymerman, J. Usefulness of diagnostic
criteria of tuberous sclerosis complex in pediatric patients.
J Child Neurol.
Oct. 2000 ;15(10):652-9.
- Jozwiak, S; Schwartz, RA; Jones, AC; Daniells, EF; Snell,
RG. et al. Molecular genetic and phenotypic analysis reveals
differences between
TSC1 and TSC2 familial and sporadic tuberous sclerosis. Human
Molecular
Genetics, 1997;6:2155-61.
- Mizuguchi, M; Takashima, S. Neuropathology of tuberous sclerosis.
Brain Dev 2001;23 (7):508-15.
- Franz, DN. Diagnosis and management of tuberous sclerosis
complex. Semin Pediatr Neurol. Dec.1998;5(4):253-68.
- Hyman, MH; Whittemore, VH. National Institutes of Health
consensus conference: tuberous sclerosis complex. Arch
Neurol. May 2000;57(5):662-5.
- Curatolo, P; Seri, S; Verdecchia, M; Bombardieri, R. Infantile spasms in tuberous sclerosis complex. Brain
Dev. Nov. 2001
;23(7):502-7.
- O`Callaghan, F; Osborne, J. Advances in the understanding
of tuberous sclerosis. Arch Dis Child 2000;83:140-2.
- Hunt, A; Sheperd, C. A prevalence study of autism
in tuberous sclerosis. J Autism Dev Disord 1993;23:323-29.
- Jones, AC; Griffiths, PD; Martland, TR. Tuberous
Sclerosis Complex: the role of neuroradiology.
Neuropediatrics 1997;28(5):244-52.
- Baron, Y; Barkovich, AJ. MR imaging of tuberous
sclerosis in neonates and young infants.
AJNR May 1999;20:907-16.
- Shepherd, CW; Houser, OW; Gomez, MR. MR findings
in tuberous sclerosis complex and correlation
with seizure
development
and mental impairment.
Am J Neuroradiol. May 1995;16(1):149-55.
- Jambaque, I; Cusmai, R; Curatolo, P. et
al. Neuropsychological aspects of tuberous
sclerosis
in relation to
epilepsy and MRI findings. Dev
Med Child Neurol 1991;33:698-705.
- Bolton, PF; Griffiths, PD. Association
of tuberous sclerosis of temporal lobes
with
autism and
atypical autism. Lancet
1997; 349:392-5.
(*) Servicio de Neurología Pediátrica
del Departamento de Pediatría del Hospital Nacional Cayetano Heredia,
lima.
(**) Genetista, Departamento de ciencias Morfolóicas, Universidad Peruana
Cayetano Heredia. |

