|
|
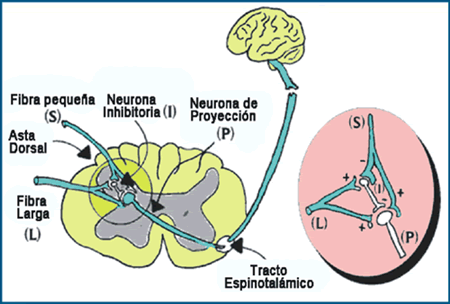
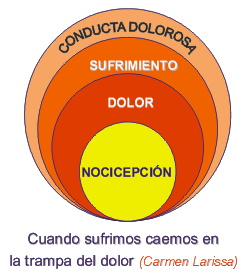
Fisiopatología del dolor Definición del Dolor Una experiencia "sensitiva y emocional desagradable, asociada a lesiones tisulares reales o potenciales, o descrita en términos de dichas lesiones". (Asociación Internacional para el Estudio del Dolor (IASP), 1986). En su perspectiva biológica el dolor permite al ser vivo una protección del daño tisular y constituye un sistema de defensa más evolucionado que el inmunológico al cual está relacionado. En general el dolor agudo vinculado a la injuria nociceptiva, estimula los receptores en las fibras dolorosas (fibra C) del sistema nervioso que lleva la información para la reacción defensiva (dolor nociceptivo). Cuando el estímulo persiste produce otra reacción inflamatoria desde el mismo sistema nervioso (neuropéptidos) que es antidrómica y autogenera un dolor crónico (regulación glial del dolor). De este modo puede iniciarse desde la misma vía dolorosa en el sistema nervioso un estímulo ó perpetuarse el mismo sin función protectora (dolor neuropático) (1). En su evolución tanto filogenética como ontogenética el ser vivo adquiere mayor capacidad de defensa que resulta en la experiencia dolorosa por la presencia del sistema nervioso. En un inicio al neuroblasto le aparecen receptores de Tirosinaquinasa-A que son comunes a otras sensaciones y desde las cuales se diferencian los receptores específicos para el dolor. Es así como el inicio de circuitos dolorosos en el recién nacido dependen de estímulos no sólo nociceptivos y es con la aparición temprana de la injuria que se vuelve específico el procesamiento del dolor (2).Finalmente de este genotipo inespecífico para las sensaciones, un fenotipo fisiológico exclusivo para el dolor con sus neuropéptidos (bradiquinina y sustancia P) aparece diferenciándose de los mecano receptores. Es así como el dolor de ser un síntoma puede transformarse en enfermedad pues su existencia ya no responde tan sólo al estímulo inicial sino que se perpetúa por lesión de la misma vía dolorosa o por sensibilización producto de la estimulación crónica (memoria sináptica) (3). Dolor Agudo Los mediadores inflamatorios tanto celulares como humorales estimularían los receptores dolorosos específicos (terminaciones libres) que a su vez desencadenan el impulso nervioso en la vía dolorosa, como consecuencia de la cascada inflamatoria (2). Por lo tanto aquí actúan todos los agentes terapéuticos anti-inflamatorios produciendo analgesia secundaria. En los receptores dolorosos se transduce el estímulo a la energía bioeléctrica del potencial de acción por transportes iónicos en los canales de Na y K a través de la membrana. El impulso nervioso se transmite por conducción centrípeta en fibras específicas del dolor amielínicas (C) y mielínicas (Alfa-Delta). Los canales de sodio en la membrana a través de los cuales se produce la avalancha de sodio son fundamentales para el potencial de acción. Hay 8 subtipos de canales de sodio que varían en su expresión genética y umbral. Sea que se origine en la injuria tisular (nocicepción) ó en la propia vía dolorosa (neurogénico) el impulso llega a la neurona del ganglio dorsal (primera neurona de la vía sensitiva), y luego hace sinapsis a través de la raíz sensitiva con las neuronas del asta posterior de la médula espinal (Figuras 1, 2 y 5) para cada segmento corporal. Desde esta segunda neurona y cruzando al otro lado asciendo por el haz espino-talámico hasta la neurona del núcleo ventro-postero-lateral del tálamo (tercera neurona). En todas las fibras y estaciones se mantiene un estricto ordenamiento topográfico para cada segmento.
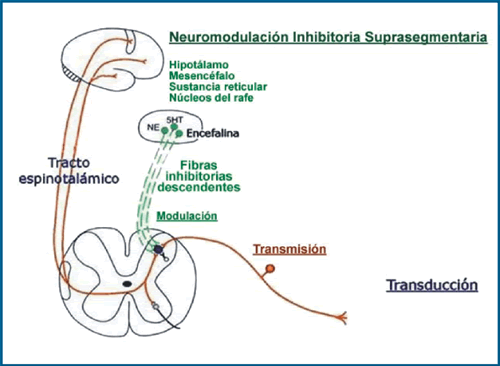
Finalmente del tálamo hay una proyección cortical vía cápsula interna a la corteza sensitiva primaria S1, S2 (homúnculo sensitivo). La entrada del impulso doloroso excitatorio segmentario (glutamato y sustancia P) es modulada por inter- neuronas inhibitorias en el asta posterior (Gaba) (4). Existe además una neuromodulación inhibitoria suprasegmentaria en todo el neuroeje. Desde el tálamo, hipotálamo, mesencéfalo (opioides); sustancia reticular (serotonina) y núcleos del rafe (beta-adrenérgicos). Llegan todos hasta la puerta de entrada del impulso a la neurona del asta posterior medular donde además la liberación de noradrenalina y acetilcolina potencian la liberación de Gaba. A su vez estos neurotransmisores de la inhibición descendente (opiáceos y serotonina) tienen receptores postsinápticos (NMDA, AMPA y Kainato) con diferentes umbrales para la entrada de sodio y potasio.
Así mismo las conexiones corticales aportan elementos emocionales primero al lóbulo límbico y luego las conexiones a otras áreas corticales aportan elementos cognoscitivos.
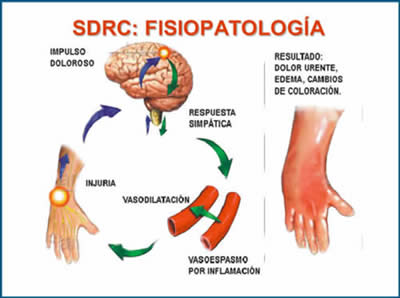
Dolor Neuropático Sensación dolorosa iniciada o causada por una lesión primaria o disfunción del sistema nervioso periférico o central. “El dolor generado por lesión primaria o disfunción del Sistema Nervioso (neuropático, periférico o central) como resultado de reacción maladaptativa en su regeneración, produce síntomas evocados y espontáneos al alterar la interpretación y equilibrio entre los impulsos dolorosos y no dolorosos”. (International Association for the Study of Pain (IASP), 1994). Dolor Neuropático Los focos ectópicos que desencadenan impulsos en la vía dolorosa en las fibras A-beta facilitando la entrada excesiva de sodio y generando parestesias y disestesias. Los impulsos ectópicos desde la fibra C también permiten excesiva entrada de sodio y sensibilización con disminución inhibitoria (Serotonina y Gaba) produciendo dolor ardiente continuo así como dolor paroxístico o lancinante. Sensibilización Periférica: el aumento de trenes de impulso incrementa los canales de sodio en la membrana de las fibras dolorosas con disminución del umbral excitatorio, tanto por su frecuencia (sumación temporal) como por su duración: Hiperalgesia. Además se producen impulsos espontáneos que aumentan la dolorabilidad. Luego por reorganización anátomo-funcional en la vía aferente (nueva sinapsis) se generan respuestas a estímulos propioceptivos provenientes de mecano receptores (fibras A-delta): Alodinia. El aumento post sináptico en la actividad de receptores de kainato (sustancia P) y entrada prolongada de calcio mantiene la despolarización que contribuye a la sensibilización de la neurona del asta dorsal. La disminución en los mecanismos inhibitorios centrales por merma en la serotonina y noradrenalina central disminuyen así mismo la actividad Gaba inhibitoria del asta dorsal (4, 6). Síndrome Regional Complejo En este caso la fisiopatogenia del dolor neuropático tiene características propias como la desproporción de la intensidad del dolor en relación a la causa así como la participación del sistema neurovegetativo creando respuestas vasculares y edema.
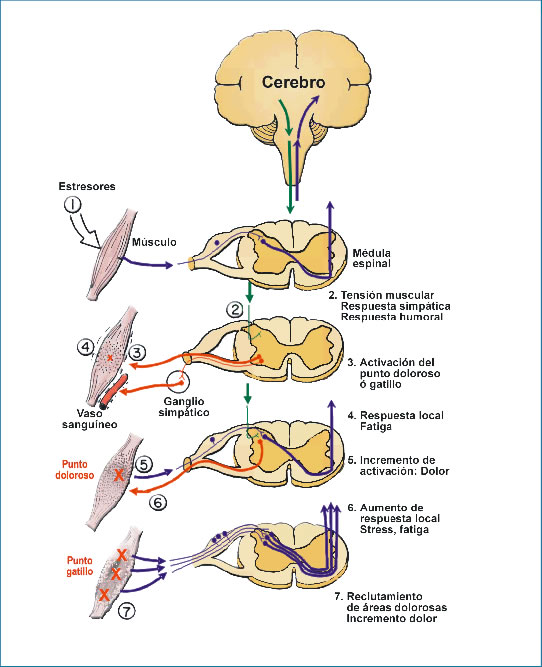
Dolor Miofascial y Fibromialgia Los múltiples factores causales involucrados tanto en el dolor miofascial como en la fibromialgia tienen componentes nociceptivos periféricos musculares y componentes neuropáticos que los llevan a la cronicidad. Posiblemente el factor neuropático sea el que contribuya a su persistencia como lo sugiere la respuesta terapéutica a los anticonvulsivantes (pregabalina) y antidepresivos (duloxetina) en el caso de la fibromialgia. En el caso de la diabetes la fisiopatogenia tiene carac-teres singulares causales (hiperglicemia) con lesión en nervio periférico que se verán en el capítulo de endocrinología (7).
Bibliografía
1 Profesor de Neurología de la Universidad Peruana Cayetano Heredia (UPCH). Médico Neurólogo del Instituto Nacional de Enfermedades Neoplásicas, Lima - Perú. |
||||||||||||||||