|
|
| Productos biológicos y biosimilares Edgardo Salinas Alva (1), Felipe Becerra Rojas (2) Introducción Un producto biológico o biofármaco es aquel que ha sido elaborado con materiales de partida de origen biológico, tales como microorganismos, órganos y tejidos de origen vegetal o animal, células o fluidos de origen humano o animal y diseños celulares (sustratos celulares, sean o no recombinantes - incluidas las células primarias) así como otros de origen biotecnológico que se obtienen a partir de una proteína o ácido nucleico por tecnología de ADN recombinante. Los productos biológicos son extremadamente más complejos que la mayoría de los medicamentos convencionales (que tienen principios activos con moléculas pequeñas (1,2). En comparación con estas pequeñas moléculas que constituyen el principio activo principal de los medicamentos sintetizados químicamente, los productos biológicos tienen un peso molecular mucho más alto y una complejidad mayor; pueden ser mezclas de muchas especies moleculares que tienen perfiles de impureza únicos, los cuales invariablemente dependen del proceso de manufactura. Por otro lado, la respuesta de los productos biológicos depende de los materiales con los que se haya comenzado su producción; Éstos están hechos a partir de sistemas vivos que inherentemente son variables. Cualquier cambio en el proceso de manufactura por menor que sea, conlleva a cambios en el producto que no necesariamente son detectables por la tecnología actual, pero pueden tener impacto potencial en la calidad, seguridad, y/o eficacia del producto. Para asegurar la consistencia en las características de los productos finales así como en los perfiles de seguridad y eficacia, tanto la fuente del material como los procesos de manufactura, la formulación y las condiciones de almacenaje deben ser cuidadosamente seguidos y controlados de acuerdo a las especificaciones que se necesitan para su aprobación regulatoria (3). Actualmente los productos biotecnológicos aceptados para su comercialización ya tratan o ayudan a prevenir: ataques cardiacos, infarto cerebral, esclerosis múltiple, leucemia, hepatitis, artritis reumatoide, cáncer de mama, cáncer de pulmón, linfomas, cáncer renal, cáncer de colon, diabetes mellitus, insuficiencia cardiaca, fibrosis quística entre otras enfermedades. Por los motivos descritos en los párrafos anteriores, se puede inferir que para efectuar el registro de productos biológicos/biotecnológicos no deberían aplicarse los criterios estándares de “Medicamentos Genéricos” (demostración de bioequivalencia con un producto de referencia mediante estudios de biodisponibilidad adecuados) tal como se lo conoce en la normativa actual de diferentes países; dichos criterios deben limitarse a los productos obtenidos por síntesis química. Aquellos que quisieran ser propuestos como “Biosimilares” o “Similares Biológicos” deberían completar un ejercicio de similaridad con el producto de referencia tanto en aspectos de calidad, como de seguridad y eficacia tanto a niveles clínicos como no clínicos. Son proteínas de elevada actividad biolígica/terapéutica, producidas en cultivos de células mediante la ingeniería genética. Cuando las células sintetizan la cadena polipeptídica inmediatamente cambian a estructuras helicoidales alfa y eso determina que se representen las actividades biológicas. Los productos biotecnológicos se diferencian marcadamente de los medicamentos de síntesis química: son de gran tamaño molecular, con pesos moleculares cien a mil veces mayores y una estructura que es notablemente más compleja. Adicionalmente presentan diferencias aún mayores: la flexibilidad de las moléculas proteicas contrasta con la rigidez de las moléculas de síntesis que no cambian su conformación (como en el caso de la Aspirina ®). De otro lado, las proteínas pueden ser modificadas por los carbohidratos (en un proceso denominado glucosilación) y por otro tipo de elementos. Los productos biológicos se diferencian de los medica-mentos sintéticos (químicos) por la complejidad de sus moléculas y los diferentes procesos que se utilizan en su elaboración. Otro elemento distintivo de los productos biológicos es que en la mayoría de casos son difíciles de caracterizar. La proteína más sencilla es compleja en si misma e incluso existen estructuras moleculares de proteínas que no pueden ser diferenciadas con la tecnología disponible actualmente. Se debe mencionar que las más modestas alteraciones en el proceso de manufactura pueden llevar a cambios significativos en un producto no detectables necesariamente a nivel de calidad de producto pero si, en el sistema inmunológico de algunos individuos (4). Parámetros como la estructura tridimensional, la cantidad de variantes ácido base o modificaciones posteriores a la traslación - como el perfil de glicosilación - pueden resultar alteradas en grado significativo por cambios que en un principio se podrían considerar “menores” dentro del proceso de manufactura (5). Se puede mencionar, entonces, que el perfil de seguridad/eficacia de los productos biológicos, depende en alto grado de la solidez y la vigilancia de aspectos relacionados con la calidad así como con los requerimientos específicos de esta industria. El desarrollo del genérico estándar - entendido como la demostración de bioequivalencia con un producto medicinal de referencia por medio de los estudios de biodisponibilidad adecuados - se debe aplicar a productos medicinales derivados de procesos químicos. En contraposición a la complejidad que revisten los productos derivados de procesos biológicos/biotecnológicos - desde el punto de vista científico - un proceso de registro abreviado sin datos propios de seguridad y eficacia, no es el adecuado. Se tendrá que adoptar un conjunto distinto de criterios clínicos para el desarrollo de un producto “biosimilar” (6,7). Los requisitos específicos para demostrar eficacia y seguridad serán diferentes para cada clase de productos; en consecuencia, el dossier de datos no clínicos/clínicos se determinará caso por caso, para aquellas situaciones en las cuales no se ha definido un lineamiento diferenciado para esta clase de productos. Debe reconocerse que por definición, los productos medicinales biológicos similares NO son productos medici- nales genéricos porque se esperan diferencias entre los productos de distintos fabricantes o en comparación con los productos de referencia, lo cual no puede evaluarse. Dada la complejidad en la elaboración de un fármaco biológico, es prácticamente imposible que otro laboratorio repita exactamente ese mismo proceso. Tanto las características del proceso como las condiciones de elaboración son patentadas y por lo tanto, de carácter reservado sólo para la compañía que lo fabrica (8). Consideraciones clínicas Es importante conocer los problemas relacionados con la seguridad de algunos productos biotecnológicos como la aparición del síndrome de Mialgia Eosinofílica por triptófano producido por cambios en el proceso de obtención biológica de este aminoácido. Hubo una impureza no reconocida que causaba este síndrome que afectá a más de 1,300 pacientes y hubo 38 muertes (1, 2). Otro caso está relacionado con la vacuna contra la encefalitis producida por picadura de garrapata. La remoción de ciertos excipientes de la formulación de la vacuna, el tiomersal y la albúmina, aumentaron marcadamente los efectos adversos de pirexia severa, escalofríos y debilidad general. La inmunogenicidad de la proteína trombopoyetina recombinante fue tan alta que provocó una trombocitopenia persistente en los pacientes que recibieron el producto, lo que determinó que se abandonara su desarrollo. Otro ejemplo de inmunogenicidad ocurrió también con el factor de colonias de granulocitos y macrófagos GM-CSF. En pacientes inmunosuprimidos fue imposible ver la inmuno-genicidad de la molécula pero al administrarla en pacientes que no estaban inmunosuprimidos, aparecieron anticuerpos contra el GM-CSF. Es importante resaltar que la inmunogenicidad no se da en todas las indicaciones y no se puede predecir. Un caso muy conocido - ocurrido en 1998 - fue la mayor incidencia de aparición de aplasia de glóbulos rojos puros (PRCA) por la administración vía subcutánea de la eritropoyetina EPREX ® dada la aparición de anticuerpos neutralizantes a la eritropoyetina (EPO) que anularon su eficacia. Este fenómeno se ha explicado debido a cambios en la formulación del EPREX ®, al eliminar la albúmina humana y reemplazarla por el Tween 80. Como consecuencia, se tuvo que retirar el producto del mercado (1). Los distintos perfiles de focos isoeléctricos en lotes de diferentes EPOs desencadenaron muchas consecuencias, considerando que estos productos trataron de copiar la formulación. Las diferencias en EPOs tienen como consecuen-cia, diferencias en el perfil farmacocinético y también un impacto en la seguridad y eficacia, ya que copiar estas presentaciones puede desencadenar reacciones adversas como inmunogenicidad. Los llamados “biosimilares” son segunda y posteriores versiones de productos biofarmacéuticos originales. Estos productos intentan tener el mismo mecanismo de acción y buscan ser utilizados para la misma indicación terapéutica del biofármaco original (9). En cambio, los biofármacos originales ó pioneros, han sido desarrollados independientemente y aprobados mediante sus propios estudios clínicos de eficacia y seguridad. No existe la probabilidad que un “biosimilar” pueda ser un biogenérico por las siguientes razones: b) El tradicional análisis químico no es suficiente para demostrar que dos productos biológicos son idénticos. Son muchas las alteraciones moleculares que pueden dar como resultado una pérdida de la actividad biológica:
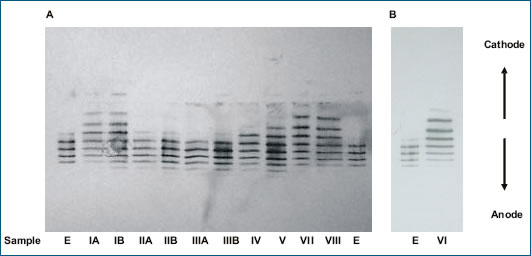
El resultado es que se producirán sustancias de diferente calidad y diferente eficacia. Mas aún, con diferentes perfiles inmunogénicos. En la figura 1 se muestra las notables diferencias entre productos biológicos supuestamente “similares” de las eritropoyetinas disponibles en Latino-América, provenientes de Corea, Argentina y Cuba:
Aunque la principal preocupación en los productos biológicos es la seguridad, es importante que tanto en los estudios clínicos como en el seguimiento de fármaco vigilancia post-comercialización se observen problemas potenciales como la inmunogenicidad y posterior falta de eficacia (10). Para un medicamento de síntesis química compuesto de una pequeña molécula, puede ser suficiente un estudio de bioequivalencia que involucre un número reducido de sujetos para demostrar su equivalencia terapéutica; pero para un producto biotecnológico y biológico complejo - es decir un biofármaco - se requiere mucho más que eso. Para establecer con un razonable nivel de certeza, que las diferencias en los procesos de producción del producto original y el similar no afectarán la seguridad y/o eficacia del producto para los pacientes, no es suficiente tener sólo la experiencia en la elaboración de dicho producto sino que se requieren los siguientes controles: • Control durante el proceso de producción (buenas prácticas de manufactura). • Estudios de inmunogenicidad, ya que pequeñas variaciones en la molécula pueden generar reacciones alérgicas severas y de consecuencias imprevisibles. • Lo más importante: estudios clínicos que demuestren la eficacia. • Fármaco vigilancia para la detección de posibles eventos adversos que esta molécula pueda generar. Todo lo mencionado, muestra que no es posible producir un medicamento bigenérico. Bajo esa consideración, existe una urgente e inmediata necesidad de establecer marcos legales que protejan a los pacientes a quienes se les administra un medicamento biológico, biotecnológico ó los llamados “biosimilares”. InmunogenicidadLos problemas de inmunogenicidad son quizá, las razones más convincentes para la imposición de pruebas clínicas humanas en los llamados “biosimilares” (11). Todas las proteínas tienen un potencial inmunogénico; esto implica la posible aparición de anticuerpos dirigidos contra las moléculas biológicas con el fin de desactivarlas ó incluso, la producción de anticuerpos que atacan el producto biológico original. De igual forma, es posible la aparición de una enfermedad autoinmune. La ruta de administración, es un factor potencial en la provocación de anticuerpos: la vía intravenosa produce menos inmunogenicidad que la ruta subcutánea ó intramuscular. Aún el mismo producto elaborado en diferentes lugares, ocasiona considerables diferencias en la inmunogenicidad - sin mostrar diferencias en las características físico-químicas. La calidad del bioproducto influencia la inmunogenicidad: un producto con grumos induce a que las células B produzcan anticuerpos, mientras que un producto soluble de alta calidad mantiene la tolerancia. Se ha demostrado, por ejemplo, que impurezas y contaminantes son la causa principal de la inmunogenicidad en la hormona de crecimiento humano y en la insulina. Las características de los pacientes también son importantes. Aquellos que padecen de cáncer tienen la inmunidad deprimida, por lo tanto, su organismo provee una menor formación de anticuerpos respecto a los pacientes con infecciones virales por ejemplo. En los pacientes hemofílicos, el tipo de defecto genético influencia en la frecuencia de inmu-nogenicidad. La importancia de la inmunogenicidad La inmunogenicidad se refiere al proceso mediante el cual, el cuerpo humano se encarga de generar una respuesta a la introducción de una proteína u otra sustancia extraña. La respuesta humana en estos casos, es producir anticuerpos que se ligan a las proteínas extrañas, desactivándolas y formando un complejo antígeno-anticuerpo que puede llevar a serias compli- caciones y efectos adversos (11). La inmunogenicidad representa la preocupación actual de seguridad más importante relacionada con los productos biológicos; principalmente se asume que es imposible de caracterizar, en ausencia de pruebas clínicas en humanos. El fabricante de un supuesto biosimilar no puede replicar jamás con exactitud el proceso seguido por el productor del fármaco biológico/biotecnológico innovador dado que las diferencias en estos procesos pueden resultar en cambios en el producto así como en sus efectos clínicos (10). Entonces, se puede afirmar que la similaridad es más difícil de establecer por los componentes bioactivos de los productos biológicos/biotecnológicos respecto a los productos obtenidos por síntesis química. Es importante precisar que se deben diferenciar las exigencias para el registro sanitario de sustancias de síntesis química respecto a las de origen biológico (12). Las agencias regulatorias más importantes del mundo actualmente se encuentran abocadas en la búsqueda de la mejor forma de normar los similares de productos biológicos origi nales. En los casos de sustancias específicas - por lo general, obtenidas por técnicas de síntesis química - no hay dificultad en acreditar la identidad y valoración del principio activo y probar que éstos sean idénticos al principio activo original de referencia. En cuanto al producto farmacéutico que contiene la sustancia química, es posible determinar si sus efectos con respecto a eficacia y seguridad son esencialmente los mismos que los del producto químico original. Para ello se usan los estudios de bioequivalencia que permiten conocer el grado de intercambiabilidad. En el caso de los productos biológicos hay dificultad para acreditar “identidad” y “valoración” del principio activo. Éstos y también los llamados “biosimilares” son producidos en seres vivos y son proteínas o anticuerpos (ambos, moléculas de altísima complejidad); por tanto, requieren validar su seguridad y eficacia en forma más rigurosa que las terapias tradicionales de síntesis química. El perfil de seguridad/eficacia de estos productos se debe demostrar en cada caso, dependiendo además de la solidez y vigilancia de su calidad - incluyendo, requerimientos de buenas prácticas de manufactura específicos para esta industria. De lo anteriormente expuesto podemos concluir que la “similitud” no puede establecerse basándose sólo en datos analíticos; es esencial, una rigurosa prueba de eficacia y seguridad con estudios clínicos y de farmacovigilancia, diseñados adecuadamente. OMS FDA (Agencia Americana para la Administración de Fármacos). Señala que los métodos analíticos no pueden predecir las propiedades biológicas de los biofármacos - El sistema inmune puede detectar alteraciones en los productos, indetectables por métodos analíticos - De otro lado, la inmunogenicidad de los biofármacos, puede tener serias consecuencias clínicas. En suma, la regulación de biofármacos no puede ser similar a la existente para las moléculas de síntesis química; por ello, la demostración in vivo de su seguridad y eficacia (a través de ensayos clínicos) es indispensable. La FDA tiene una estructura propia para el registro de productos biológicos y documentación especifica - NO existe un procedimiento abreviado para el registro de biofármacos;por lo tanto, no tienen un procedimiento normativo para el registro de medicamentos biológicos similares. EMEA (Agencia Europea para Evaluación de Productos Medicinales). Entidad avanzada en el establecimiento de estándares científicos para aprobación de este tipo de productos. Desde Octubre 2005 ha elaborado lineamientos sobre “Productos medicinales Biológicos similares”. EMEA es muy estricta en sus guías para biosimilares y exige: • Información completa sobre química, fabricación y control de calidad. • Extensos estudios de comparabilidad entre “biosimilares” y “biofármacos de referencia”. • Ensayos clínicos de extensión según el producto (caso a caso). • Investigación clínica en cada nueva indicación. • Estudios clínicos que deben incluir diversas pruebas como la de inmunogenicidad y la de farmacovigilancia así como el compromiso post-marketing correspondiente. En tres países, se ha logrado un marco regulatorio diferenciado para el registro de Biofármacos, así (2): • Brasil: ANVISA (Agencia Nacional de Vigilancia Sanitaria) tiene una regulación específica para biológicos que exige la presentación de sus propios estudios clínicos. • Venezuela: El INHRR (Instituto Nacional de Higiene “Rafael Rangel”), tiene una norma que regula la exigencia de ensayos clínicos para biológicos/biotecnológicos. Clasifica los productos en “recombinantes” y “anticuerpos monoclonales”. La norma hace énfasis en fabricación y control. • Chile: El ISP (El Instituto de Salud Pública) recientemente ha regulado un sistema especial de registro de Biofármacos que incluye ensayos clínicos de eficacia y seguridad asi como farmacovigilancia post marketing. • Colombia: INVIMA (Instituto Nacional de Vigilancia de Medicamentos y Alimentos) no tiene una normativa específica para registro de productos biológicos, biotecnológicos ni biosimilares; sólo en casos excepcionales se han solicitado estudios clínicos propios con la molécula para aprobarla como biosimilar (eritropoyetina, inmunoglobulina, heparina, soma- totropina e insulina). • México: No existe una normatividad específica para los productos biotecnológicos. La industria farmacéutica ha sugerido que en todos los casos se debería cumplir con pruebas clínicas para demostrar eficacia y seguridad en virtud que - debido a los estándares de producción y/o fabricación - no se pueden considerar los datos de prueba del producto original con el supuesto biosimilar. No obstante, se han otorgado registros de “biogenéricos” sin existir una normativa específica. En base a las experiencias anteriormente relatadas, se puede concluir que cualquier marco regulatorio para la aprobación de biofármacos debe:
Conclusiones
Bibliografía
1 Oncólogo Clínico del Hospital Edgardo Rebagliati Martins - EsSalud, 2 Médico especialista en Inmunología y Reumatología, |
||||||||||||||||||||||||||