|
|
| Síndrome de Sturge Weber:Reporte de un caso y revisión de la literatura
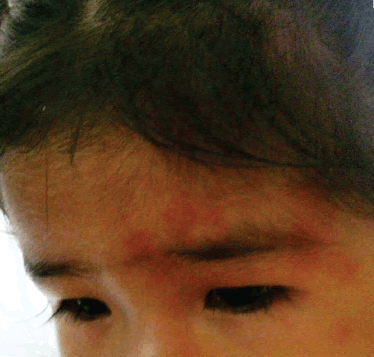
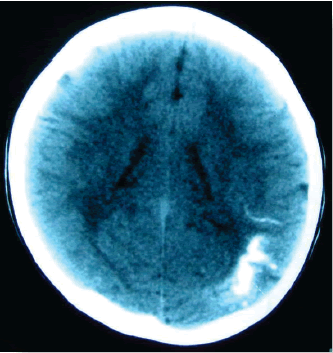
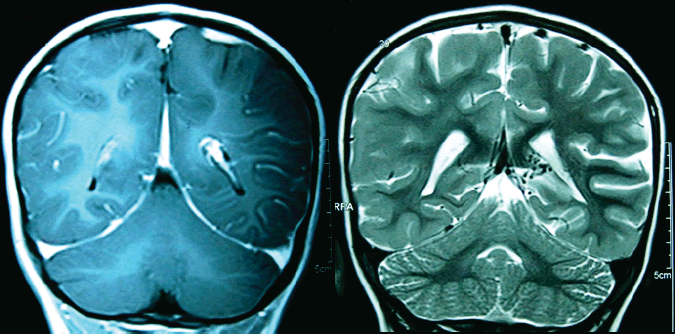
Erik Guevara Silva (1),María Garay Egashira(2) IntroducciónIntrodución: El síndrome de Sturge-Weber (SSW) es una entidad neurocutánea congénita caracterizada por una malformación vascular facial (mancha en vino de Oporto) asociado a angiomatosis leptomeníngea. Su prevalencia es aproximadamente de uno por cada 50 000 nacidos vivos. Afecta por igual a ambos sexos. Las manifestaciones clínicas incluyen a las convulsiones, nevus vascular cutáneo unilateral en relación a la división oftálmica del nervio trigémino, glaucoma ipsilateral, hemiparesia contralateral, hemiatrofia, hemianopia y retraso psicomotor. La característica radiológica es la presencia de calcificaciones giriformes en los lóbulos occipital y parietal. Reporte de caso: Una paciente de 1 año 7 meses llegó a la emergencia presentando convulsiones generalizadas que cesaron con diazepan, y se inició tratamiento con ácido valproico. Mostraba una mancha congénita color vino de Oporto en la frente, además se evidenció un leve retraso psicomotor. La tomografía mostró una calcificación cortical en los lóbulos parietal y occipital izquierdos. En la resonancia magnética se evidenció un reforzamiento leptomeníngeo parietal izquierdo. Conclusiones: El SSW es una entidad rara y de curso progresivo, el diagnóstico no es difícil cuando las manifestaciones típicas están presentes. Una malformación cutánea facial y la presencia de retardo mental debería alertar a los médicos cuando se encuentran frente a niños con convulsiones focales o parciales complejas secundariamente generalizadas en el primer o segundo año de vida. Palabras clave: Síndrome de Sturge-Weber, retraso psicomotor, convulsiones infantiles, facomatosis, malformación facial cutánea. IntroducciónIntroduction: Sturge-Weber syndrome (SWS) is a congenital neurocutaneous syndrome characterized by unilateral facial cutaneous vascular malformation (Port-wine stain) in association with ipsilateral leptomeningeal angiomatosis. Prevalence is approximately one per 50 000 live births. Males and females are equally affected. Clinical manifestations include seizures, unilateral cutaneous vascular nevus following the ophthalmic divisions of the trigeminal nerve, ipsilateral glaucoma, contralateral hemiparesis, hemiatrophy, hemianopia and psychomotor retardation. The radiographic hallmarks of SWS are gyriform calcifications usually involving the occipital and parietal lobes. Case report: A 1 year and 7 month-old girl came to emergence department showing generalized seizures that resolved with diazepam and valproic acid was started. She had a facial Port-wine birthmark located on the forehead; a mild psychomotor retardation was evidenced. Head CT demonstrated a cortical calcification of the left parietal and occipital lobe. MRI showed a left parietal leptomeningeal enhancement. Conclusions: SWS is a rare and progressive entity, the diagnosis is not hard when typical manifestations are present. Mental retardation and facial cutaneous malformations should alert physician when they are in front of infants with focal or complex partial/secondarily generalized seizures in the first or second year of life. Key words: Sturge-Weber syndrome, psychomotor retardation, infantile seizures, phacomatosis, facial cutaneus malformation. IntroducciónEl síndrome de Sturge-Weber o angiomatosis encéfalo-trigeminal es una enfermedad rara que pertenece al grupo de las facomatosis, de carácter congénito y no hereditario. Se caracteriza por la presencia de una malformación vascular cutánea facial denominada mancha en vino de Oporto, en asociación con angiomatosis leptomeníngea ipsilateral. También forman parte de este síndrome las crisis epilépticas, retardo mental, hemiparesia contralateral y glaucoma ipsilateral (1). Esta patología constituye el cuarto síndrome neurocutáneo más frecuente después de la neurofibromatosis tipo I, esclerosis tuberosa y la hipomelanosis de Ito. La prevalencia es aproximadamente un caso por 50 000 nacidos vivos (2,3). El síndrome de Sturge-Weber es un diagnóstico infrecuente en nuestro hospital y puede pasar desapercibido en parte por la carencia de exámenes auxiliares que se requieren para su confirmación, la importancia del diagnóstico de esta enfermedad crónica y progresiva radica en elegir el mejor tratamiento para el paciente y poder informar mejor a la familia pronóstico y curso de la enfermedad. Todo esto nos motiva a la presentación del siguiente caso. Presentación de casoPaciente mujer de 1 año y 7 meses de edad sin antecedentes perinatales ni familiares de importancia, con inmunizaciones completas, que recibió lactancia materna exclusiva hasta los 6 meses. Ingresa por el servicio de emergencia por presentar crisis epilépticas que inician con oculocefalogiria hacia la derecha seguido de movimientos tónico-clónico generalizados, la paciente llegó despierta pero durante su estancia en emergencia presentó una crisis de las mismas características que se controló con diazepan endovenoso y se inicia valproato de sodio 10mg/kg de peso con aumento progresivo en los siguientes días, no presentó fiebre ni vómitos, la paciente fue hospitalizada para mayor estudio de la etiología de las convulsiones. Al examen físico se observó una mancha cutánea eritematosa congénita, plana, de bordes irregulares, sin presencia de vello cutáneo, localizada en la región fronto orbitaria izquierda (Figura 1). Al examen neurológico, paciente se muestra activa, adecuada reactividad a estímulos, fuerza y tono muscular adecuados, no signos meníngeos. Se evidenció un leve retraso psicomotriz principalmente en el área del lenguaje (solo llega a decir hasta cinco palabras). La evaluación oftalmológica no mostró alteraciones. Los exámenes de laboratorio fueron normales. La tomografía cerebral mostró calcificaciones en la región parieto-occipital izquierda (Figura 2). En la resonancia magnética se observó refuerzo leptomeníngeo parietal izquierdo con hipertrofia del plexo coroideo característico del angioma, sin atrófia cortical, hallazgos compatibles con las manifestaciones tempranas del síndrome de Sturge-Weber (Figura 3). No se realizó electroencefalograma por no contar con este estudio en el hospital.
Este síndrome fue descrito por primera vez por Schirmer en 1860, y fue Sturge quien efectuó su descripción clínica completa en 1879; posteriormente Weber, en 1922, demostró las alteraciones radiográficas típicas de la enfermedad (4). El síndrome de Sturge-Weber es una facomatosis cuya etiología no está claramente determinada que cursa con manifestaciones neurológicas, oftalmológicas y dermatológicas. La principal anomalía vascular intracranial es la angiomatosis leptomengíngea que puede afectar a uno o ambos lóbulos. Aunque la mancha facial es una lesión relativamente común, esta ocurre solo en 3 de cada 1000 nacidos vivos y solo el 5% de los infantes con esta malformación tienen la enfermedad. Por el contrario, el 13% de los pacientes con manifestaciones cerebrales no muestran el nevus facial (2). Entre el 75% al 90% de los pacientes presentan convulsiones focales o generalizadas las cuales frecuentemente se presentan en el primer año de vida y pueden progresar hasta llegar a ser refractarias a la medicación y desarrollar una hemiparesia permanente o transitoria. Así mismo, alrededor del 70% de casos desarrollan retraso psicomotor en diferentes grados y requieren educación especial, esta complicación es menor en aquellos que no desarrollan epilepsia (2,4,6).
Las anormalidades encontradas en esta patología pueden explicarse desde las malformaciones del plexo vascular embrionario, surgiendo dentro del mesénquima cefálico entre el neuroectodermo y la vesícula telencefálica; la interferencia del drenaje vascular en estas áreas aproximadamente entre la quinta a octava semana de gestación posteriormente afecta la cara, ojos, leptomeninges y el cerebro. La mancha facial congénita es una malformación capilar que consiste en capilares congestionados en el tejido subepidérmico por involución incompleta de la vasculatura embriogénica. Los estudios radiológicos e histopatológicos del tejido nervioso demuestran la evolución progresiva de las calcificaciones cerebrales y su origen vascular; esto es probablemente debido a la permeabilidad de las paredes de los vasos y al estancamiento de la sangre en estos vasos, ocasionando anoxia del endotelio. La angiomatosis se acompaña de un pobre drenaje venoso cortical con dilatación venosa, el plexo coroideo ipsilateral también puede llegar a dilatarse Por lo tanto las lesiones características son el refuerzo leptomeníngeo de la angiomatosis, dilatación venosa, agrandamiento de los plexus coroideos, anormalidades en la sustancia blanca, atrofia y calcificaciones corticales (2,8,9). El tratamiento de esta enfermedad es multifactorial y multidisciplinario. En los pacientes con crisis bien controladas y un desarrollo psicomotor normal o casi normal, el tratamiento es médico y conservador. Es importante el diagnóstico temprano de esta patología para un mejor control de las convulsiones con fármacos como fenitoína, carbamazepina, ácido valproico, levetiracetam, entre otros; se prefiere iniciar con monoterapia y asociar una segunda droga en casos de refractariedad, la opción quirúrgica se reserva para aquellos casos en que ninguna de estas drogas funcionen. Está indicada la medición periódica de la presión intraocular debido al riesgo de padecer glaucoma La estimulación temprana, terapia psicomotora y terapia de lenguaje son aspectos importantes en el tratamiento de estos pacientes. Las lesiones faciales pueden ser tratadas con cirugía o laserterapia (2,10,11,12). Estudios en poblaciones pequeñas han sugerido que el ácido acetilsalicílico a dosis bajas puede reducir el riesgo de enfermedad cerebrovascular (8) así como la frecuencia y severidad de las convulsiones (3,12). Nuestra paciente muestra las lesiones dermatológicas y neurológicas compatibles con el síndrome de SW, la presencia de convulsiones en una paciente con mancha eritematosa facial plantearon a esta enfermedad como una de las primeras posibilidades diagnósticas, posteriormente los estudios de neuroimagen nos ayudaron a confirmar nuestra sospecha inicial. Actualmente las convulsiones están controladas con ácido valproico, también se ha indicado ácido acetilsalicílico y la paciente está recibiendo terapia de lenguaje y de estimulación psicomotriz. Se ha sugerido a los padres la inmunización contra la influenza debido a que los pacientes que reciben ácido acetil salicílico y adquieren esta infección podrían desarrollar el síndrome de Reye. Finalmente, en este caso la enfermedad no se ha presentado con la severidad que describe la literatura; sin embargo, los padres fueron informados del posible curso de la enfermedad. En conclusión este caso sugiere que en un paciente pediátrico que debuta con convulsiones y lesiones dérmicas faciales congénitas debe plantearse el síndrome de Sturge Weber como uno de los diagnósticos diferenciales. Referencias Bibliográficas
1 Médico Neurólogo. Departamento de Medicina del Hospital Regional de Huacho. Universidad Nacional Mayor de San Marcos (UNMSM). Lima-Perú. Investigador Principal de la ONG Inmensa.2Médico Pediatra. Departamento de Pediatría del Hospital Regional de Huacho. Universidad Federico Villarreal. Lima-Perú. |
||||||